药物化学
摘自:郭保章《20世纪化学史》;442-474
第十二章 药物化学(442-)
药物化学是有机化学的重要分支。由于有机化学中各分支以及生物化学、生理学、药理学等的发展,以及计算机、生物技术等在药物化学中的应用,近年来,药物化学有了很大发展,而且文献浩繁。此处只准备就20世纪药物化学发展几个重要阶段进行讨论,中草药不在讨论之列。
药物对增进人民健康的作用是不言而喻的。在美国,平均估计寿命从1900年的49岁上升到1976年的73岁,而预期到2000年将超过79岁。主要起作用的因素是普遍应用各种各样的新型医用化合物。
在过去几十年间药品柜里的东西发生了巨大的变化。对第一次世界大战前夕的内科医生进行的调查揭示出,当时最基本的10种药品是乙醚、鸦片及其衍生物、毛地黄、白喉抗毒素、天花疫苗、汞、酒精、碘、奎宁和铁剂。在第二次世界大战结束时又做过一次调查,列在常用药品表首位的是磺胺药、阿斯匹林、抗菌素、血浆及其代用品、麻醉剂和鸦片衍生物、毛地黄、抗毒素和疫苗、激素、维生素和肝浸膏。我国也有类似的情况,在六七十年代,有人形象地把内科医生处方称为“潘金莲”(盘尼西林、金霉素、链霉素字首的谐音)。今天有更多种医用化学品,但是退热药、止痛药和消炎药在医疗实践各个领域的药品表中仍居首位。每年都要试验许多新药,但真正能投放到市场上出售的却很少。
一、化学药物的开端(443-447)
19世纪末叶出现了一股寻找具有药用价值化学品的热潮。P・艾利希(Paul
Ehrlich,1854-1915)是化学疗法的最热情的探索者之一。虽然他所作的研究工作可以追溯到19世纪70年代,但他所作的主要贡献却在20世纪才应用起来。当艾利希在布雷斯劳和莱比锡还是一名学生的时候,就对染料及它们对于活组织的作用产生了极大的兴趣。他的表兄C・威格特(Carl Weigert,1845-1905)教了他给细菌染色的技术。威格特并不是第一个把有色化合物作为生物着色剂的人;1875年在威格特用甲基紫着色显示动物组织内的细菌之前,人们将铬酸、洋红和苏木精用于同样的目的作法已有1O年以上的历史了。各种不同的染色法在细菌学家和组织学家中间迅速地发展起来了。R・科赫(Rober Koch,1843-1910)把细菌对染料的反应作为辨别不同细菌的工具,H・C・J・格拉姆(Gram)在l884年介绍了他所采用的鉴别技术,其他许多科学家作出了进一步的贡献。
基于一些具体的染料对某些细菌或某种组织的选择性,艾利希(EMich)得出这样一种见解:使用恰当的染料来治疗疾病应该是可能的。他于1887年证明了亚甲基蓝可使活神经细胞着色但不影响邻近的组织,同样地它能使某些细菌着色而对其它细菌无影响。能查找出某些染料,它可以附着在某一特定的生物体上,从而将该生物体杀死而又不损害宿主生物体细胞呢?
l889年艾利希成为了罗伯特・科赫在柏林的传染病研究所的成员。1892年当E・冯・贝林(Emil.Von Behring,1854-1917)发现医治白喉的抗毒素时,他已与E・冯・贝林建立了密切联系。艾利希同血清的发展有很大的关系。他后来成为了在美因河畔法兰克福的国家血清研究所的所长。虽然他整天忙于血清生产和实验,他仍继续努力去寻找一种既对病原体有高度专一性而对高等动物又相对无毒的染料。他得到卡西乐化工厂的合作,为他提供在他们实验室里生产出来一些新化合物样品。此外由于乔治・施派尔-豪斯公司(Geong
Speyer-Haus)在1906年建立,他得以置身于一群助手――化学家们和细菌学家们中间,进行化合物的合成和改性工作,并研究这些化含物对于病原体和动物的效应和作用。
在早期阶段艾利希提出了他的具有杀菌作用的侧链理论。根据这一理论设计一种具有侧链的分子而且对某一寄生菌有互补作用应该是可能的。用侧链的方法把这种分子附着到微生物体上。可能阻碍微生物的活动或者可能杀死微生物。既然这些侧链将只会作用于病原生物体而不会伤害宿主细胞,因此设计这些有效的魔弹将是可行的。他的这些想法部分是受到血清疗法的成功的影响而产生的。在这里病原体本身刺激着那些使病原体死亡的特别活跃的物质的形成,而不伤害大多数宿主细胞。因为不可能造出许多有效的血清来治疗多种疾病,所以有必要发展化学疗法,制造出使寄生细菌致死的具有特效的化合物。
因为对一种生物体来说是有毒的某一化学药品差不多肯定会对其他细胞显示毒性,艾利希提出了治疗指数(therapeutic index)作为保证化学药品使用的安全标准,该项指数实质上是宿主动物所能忍受的最高剂量与有效治疗剂量之比。
早在20世纪初艾利希和志贺(清).(洁)就发现锥虫红对治疗锥体虫所引起的疾病有特效。而F・E・P・梅斯尼尔(Mesnil)和M・尼科尔(Maurice Nicolle)则证明了锥虫蓝的疗效则更高。这些药物在治疗一些热带疾病如昏睡病和马锥体虫病方面取得了某些成功。1906年科赫(Koch)采用对氨基苯胂来治疗人类由锥体虫引起的疾病。该化合物是由贝尚(Bechamp)于1863年制备成功的,据信是砷酸的酰替苯胺。 1905年利物浦的医生H・W・托马斯和A・布列恩尔(Breinl) 在也们的报告中宣布该化合物具有毒杀锥体虫的作用;所以用对氨基苯胂(Atoxyl无毒的)这一名称,因为它对宿主并无毒性。艾利希知道砷和氮在周期表中属于同一族元素,因此对砷化合物极感兴趣;他征实了该化合物对锥体虫有作用,但发现它不能使用,因为它的毒性太强会损害视觉神经。他对贝尚所制备的对氨基苯胂的化学结构,表示怀疑,并提出了该化合物的正确结构。因为他对染料有丰富的实践经验,所以他还提出,该结构会出现一个游离的氨基。
大约在这一时期,引起梅毒的生物体梅毒螺旋体(Treponema pallidum)被E・霍夫曼(Hoffmann)和F・绍丁(Fritz Schaudinn)发现。绍丁指出,该生物体(一种螺旋体)与其说是细菌,倒不如说它更具有原生动物的特征。艾利希设计了能在患这些疾病的兔子和老鼠身上作试验的一些新的砷化合物。从偶氮基在锥体红这样的染料中是一个有疗效的单元这一点出发,他运用推理方法假定三价砷可能比五价砷更为有效,正如在对氨基苯胂中所发现的一样。他的化学师A・伯塞姆(Alfred
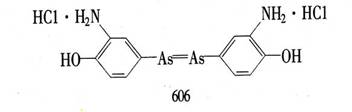
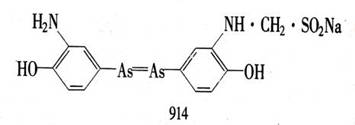
Bertheim)制备了这些化合物;发现阿撒司丁(对乙酰胺基苯胂酸)是治疗锥虫病实验的特别有效的物质。第418号化合物,即胂苯甘氨酸被证明更为有效。更多的化合物在实验室里被合成出来,并在他的日本助手羽田佐八城(Sahachim Hata)的指导下进行了试验。1909年用606号化合物治疗梅毒获得成功。该药物后来在市场上销售时名为洒尔佛散或胂凡纳明。后来在1912年又有一种更方便的化合物914――新洒尔佛散被研制出来了。使用这些砷制剂的治疗法很快就被介绍开来。虽然新洒尔佛散并不是没有缺点,但40年代前,在有效的抗生素仍未被用来治疗梅毒的时期内,它一直是医治这种疾病的标准药物。
|
|
|
|
含砷制剂成为特效药这一成功给人们带来了一种极乐观的情绪。认为化学工业可以制造类似的各种化学治疗药剂。然而一切热情又变成了失望。因为对那些有疗效的化合物的研究结果并不能使人知道应该如何合成各种分子,使之对某些疾病既有特效而又不会对宿主有害。除了拜尔205和几样其它化合物外,在1910年-1930年之间在研制化学治疗剂方面几乎没有获得真正的成功。
拜尔205即日耳曼宁,在1920年被采用为治疗非洲昏睡病的特效药。日耳曼拜尔公司(在第一次世界大战中这家公司的美国的分支机构被查封并被迫改为一个独立的公司)在这些药品被严格限制的情况下弄到这些药品,因而被指控为一直利用药品作为一种政治武器来帮助德国收复失去的殖民地。巴斯德研究所(Pasteur Institute)的E・福尔诺(Ernest Foumeau,l872-1949)尽管是在末获得专利权和拜尔公司拒绝为他的研究提供药品的情况下,还是把这种化合物鉴定出来了。一群科学家连同不列颠染料公司在解决上述的困难题中也起了积极的作用。通过查阅战前德国的专利文献,福尔诺得知他们对复杂的尿素衍生物进行了大量研究。在这些公开专利文献里还有证据进一步表明:目前对于由一些酰胺键合氨基苯甲酰基和萘胺苯磺酸端基有很大兴趣,就像对锥虫染料的兴趣一样。通过一种测试分析排除了几百种可能的化合物之后,范围缩小到25种,并对每种化合物都进行了合成以及生物和化学方面的实验。其中的一种福尔诺309被证实具有拜尔205所具有的杀锥虫能力,无毒性并具有化学稳定性。与巴斯德研究所设法弄来的56mg德国产品相比较的结果证明,两种化合物是完全一样的,德国人拒绝承认这两种化合物是一样的;福尔诺309在英国和美国获得专利权,该化合物对早期昏睡病有显著疗效,但对晚期昏睡病却无能为力。1919年W・A・雅各布斯(Walter
Abraham Jacobs)和洛克菲勒医药研究所的M・海德尔伯格(Michael Heidelberger)证实了锥虫砷胺对影响中枢神经的系统的晚期昏睡病具有疗效。
二、抗疟剂(447-448)
19世纪末,抗疟疾药物得到了极大的重视,虽然喹啉早已被证明是构成奎宁分子的要素,但许多年内奎宁的剩余部分却一直被认为是“次要的另一半”。在19世纪末20世纪初之间,斯克劳普(skraup)、柯尼希(Konigs)和拉贝(Rabe)正积极从事于用这“次要的另一半”来鉴别降解产品的工作。这些产品被称为洛滂(1oipon)或梅罗斯(meros)。这另一半的结构慢慢被弄清楚了;1913年P・拉贝(Paul Rabe,1869-1952)从中探得了一种合成物,最后在1944年伍德沃德(Woodward)和多林完成了奎宁的全合成工作。
与此同时许多化学家们正在他们的实验室里进行寻找一种合成的代用品。l926年I・G・法本公司制出了扑疟喹啉(Plasmochin)。该药物能杀死疟疾原虫的抗奎宁生殖体,但由于其毒性太强不能广泛使用。阿的平大约在1903年被应用,但直至第二次世界大战中爪哇的奎宁供应被切断后,这种药物才被普遍使用。它像奎宁一样能侵袭处于裂殖体时期的寄生虫,并能防止对红血球的破坏(红血球被破坏是冷热病的原因)。它是一种吖啶型染料,会引起暂时性黄色素沉降。由于其来源广,效果佳,它现在仍然被使用着。
由于战争初期抗疟疾药物的短缺,美国实行了一项雄心勃勃的研究计划,要将所有可能成为抗疟疾药剂的各种化合物进行合成和检验。但检验结果除了在l4000种可能性较大的化合物中发现出一定数量的更具有可能性的化合物外,无一能够作为阿的平和奎宁的合适代用品。抗日战争期间,大后方昆明的条件十分困难,26岁的年轻化学家邢其毅为了寻找抗疟药物,千方百计收集云南边境地区的金鸡纳树种,开展对我国河口金鸡纳的成分分析提纯研究,并取得成果。
此外在抗疟药物方面,中国科学工作者曾调查分析出多种抗疟中草药,其中有常山和青蒿,效力超过奎宁。90年代中国科学院上海药物研究所新药研究国家重点实验室研究员朱大元等10位科学家在研究青蒿素及其衍生物合成中做出杰出贡献。
三、磺胺类药物
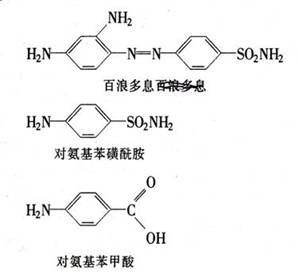
磺胺类药物是在30年代开始应用的;最先被使用的磺胺药物是I・ G・法本公司的百浪多息,由G・多马克(Gerhard
Domagk,1895-1964)开始用于治疗链球菌和葡萄球菌感染的动物试验。多马克是一位受过专业培养的内科医生,从探索某些染料应用于医学上的可能性这样观点出发,开始对染料进行系统的研究。当时有一种新合成的染料,这是一种桔红色的化合物,其商品名称为百浪多息。1932年,多马克发现注射这种染料对老鼠的链球菌感染非常有效。
这是一项使人极为兴奋的发现,大约30年前艾利希和其他一些人已经发现了若干疾病的化学治疗剂,不过这些疾病,像锥虫病是由原生动物引起的;或者像梅毒,则是由不常见的细菌造成的。对于更常见而微小的细菌则纯化学性质的治疗还无能为力。
多马克通过非常直接的途径发现百浪多息的作用对人类也是适用的。他的小女儿因为被针刺了一下而受到链球菌的感染。在采用多种方法治疗无效后,多马克在绝望中对他注射了大剂量的百浪多息。她很快地恢复了健康,因而在1935年世界上知道了这种新药。
当百浪多息被用来挽救美国总统的儿子――小F・D・罗斯福时,这种新药便获得了更大的名声;当时这位病人也是因为感染而生命垂危。英国首相温斯顿・邱吉尔也是因为受到百浪多息的治疗而恢复健康的。 1939年,多马克由于自己的发明而光荣地获得了诺贝尔医学或生理学奖。
这个工作后来在全世界的研究实验室继续进行,30年代初期连亚历山大・弗莱明都把青霉素放置一旁而去研究百浪多息。但是最有意义的结果是由巴斯德研究所的丹尼尔・博维特(Daniele Bovet,l907-)取得的,博维特认识到并非所有百浪多息分子都是获得明显抗菌作用所必需的。博维特和他的同事们对这种红色染料进行了实验,发现该化合物虽然对试管内的细菌毫无杀伤作用,但在人体组织中却能分解成为对氨基苯磺酰胺。福尔诺(Foumeau)发现对氨基苯磺酰胺具有与百浪多息相同的效力,并且该药物很快投入使用。该药物没获得专利权因为它是P・盖尔莫(Gelmo)早在1908年制成的。I・G・法本公司后来被指控在知道了这种化合物的疗效之后却在其结构的第一环上加上别的基,并利用其专利权,使人们无从懂得如何合成该药物。
|
|
对氨基苯磺酰胺的研制成功,引起了人们对其它各种含磺酰胺基的化合物进行检验的热情。五年之内各制药公司对上千种以上的化合物进行了合成和测试工作。发现一小部分化合物具有治疗价值,并且很快就被应用于医疗实践。
剑桥大学的两位生物学家P・法尔兹(Paul Fildes)和D・D・伍兹(D.D.Woods)对磺胺类药物作用的方式提出非议。根据他们的观察,在试管内对氨基苯磺酰胺对于从不同的培养基培养出来的生物体具有强弱不同的效果。酵母提取物含有抑制药效的的物质。当培养基含酵母提取物时需要更大量的磺胺来杀死细菌。这种作用与把琥珀酸脱氢为富马酸的过程中,丙二酸对琥珀酸脱氢酶所起的抑制作用是相似的。
伍兹发现酵母提取物所含的对氨基苯(甲)酸(PABA)与具有对抗作用的对氨基苯磺酰胺极为相似。这种对抗作用发生在生物体需要对氨基苯(甲)酸的生物新陈代谢过程中。几年后,在对叶酸的研究中发现,对氨基苯(甲)酸是叶酸分子的一个组成部分。当存在大量的对氨基苯磺酰胺时,那些本身具有合成叶酸功能的细菌造酸功能就受到阻碍。某些无害的细菌,还有高等动物需要从食物中摄取现成的叶酸,因而不会受到对氨基苯磺酰胺的伤害。
法尔兹提出,设计某些与新陈代谢所需的物质相似的药物应该是可能的。于是很多化学工作者公开了这样一些对抗药的作用Pyrathiame能抑制那些不能合成维生素B1的生物体的生长,泛磺酸对那些需要泛酸的生物体显示一种类似的对抗作用。总的来说这次对化学疗法的探讨并没有取得惊人的进展,因为微生物有许多获得营养的本领,而我们对于它们新陈代谢过程仍然是所知极少。接着面临的问题在于使用某种药物后,经常出现在某种细菌身上不断增强的免疫力。这是艾利希早已意识到的问题。虽然很容易把易受影响的菌株杀死,但具有抵抗能力的细菌能生存下来并取得优势。
四、抗菌素(451-454)
磺胺类药物标志着在化学疗法方面的一大突破,但在40年代它却在很大程度上被迫让位于抗菌素。虽然霉的抗菌作用曾被报导,而且对霉的作用做了至少好几次的调查研究,直至1928年A・弗莱明(A1exander Fleming,1881-1955)出名后,这类药物才开始它自己的历史。有一次他在度完小休之后回到自己实验室时,观察到在一只培养葡萄球菌的培养皿上生长了一种蓝色霉菌。在这种霉菌周围有一晕圈,在晕圈内,没有细菌生长。这种霉菌显然分泌出一种致死物质通过培养基向外扩散,它们所到之处细菌都被杀死。弗莱明知道了这种培养基含有抗菌物质并命名为青霉素,这种霉对实验动物不显示毒性。
由于弗莱明在伦敦的圣玛利医院供职,医务缠身,使他不能专心致志地继续观察研究;而随着磺胺类药物的出现及使用效果使人们普遍对他关于青霉素的报告不感兴趣。直至1936年在澳大利亚出生的H・W・弗洛里(Itoward Walter Florey)和从纳粹统治下逃出的难民E・B・钱恩((Ernst Boris
Chain)才在牛津重新对青霉素进行实验。他们证实了弗莱明的观察结果并继续工作,从某种不纯净的药粉中提取一些物质。这些物质对于被实验动物身上某些细菌具有特别强的杀伤力。1942年化学家钱恩制成了一种纯净的化学药粉。弗莱明把这种药粉用来治疗脑膜炎并且取得了成功。这时弗洛里访问了美国,知道在联邦政府的帮助下,默克、普菲泽尔和斯奎布等公司已制订了进行工业生产计划。联邦政府的雄厚资金给这项研究和生产以有力的支持,及时解决了青霉素面临的生产问题,使之能为战争提供充足的药品。
对于青霉素的化学研究人们也进行了相当大量的工作。不久英国和美国的化学家们的研究结果证实了有好几种青霉素的存在。根据D・C・霍奇金(Dorothy
Crowfoot Hodgkin)的研究结果,人们弄清了肯霉素的结构。发现不同的青霉素具有大致相同的结构,差别只是在于某一侧链的结构不同。不久人们也清楚地知道了,合成这种分子是不容易的。但是用生物合成的手段使合成各种各样的青霉素获得惊人的成功。1957年J・C・希恩和K・R・亨纳利洛根经过了九年的研究之后,合成了青霉素V。
大约从1940年开始,人们对于抗菌素的兴趣越来越浓。越来越多的人从事于由微生物产生出来的抑制细菌物质的基础研究。 1939年R・J・杜博(Rene J.Dubos,1901-?)这位一直在洛克菲勒研究所工作的学者宣布,他从短杆菌中分离出一种抗菌的物质。这种物质不久就被分成短杆菌酪和短杆菌肽。虽然它们以结晶状被提取出来,但它们后来都被分离为各种相应的多肽。 1944年苏联的高斯(Gause)所宣布的短杆菌肽S也是一种多肽。虽然这些化合物对于革兰氏阳性细菌具有药效,但由于它们的毒性强,使它们不能作为药物来使用。
瓦克斯曼(Waksman)和他在鲁特格斯(Rutgers)的同事们共同研究,在1943年发现了链霉素. S・A・瓦克斯曼(Selman.A.Waksman,1888-1973)是第一流的土壤细菌学家,他从一种土壤生物体中分离出灰色链霉素这种物质。链霉素是一种治疗肾脏传染病、肺结核病和其他好几种青霉素不能治疗的疾病的一种有效药剂。要证实这种物质的复杂化学结构需要大量的研究。它是由一个碱基(链霉胍),一个糖(链霉糖)和一个氨基葡萄糖的残基构成的。在天然糖的衍生物中这种葡萄糖的残基是唯一显示L构型的.
早在40年代,许多制药厂对世界所有的由土壤微生物制成的药物进行了检验,竭力从中找出一些新的和有用的抗菌素. P・戴维斯(Parke Davis)所发现的氯霉素,就是从委内瑞拉土壤标本里得到的一种霉(放射线菌属委内瑞拉链霉菌)中获得的。这种化合物结构简单所以它很快就被用合成法制造出来在市场上销售了。
金霉素是由B・达格尔(Benjamin Duggar,1872-1956)于1948年化里德尔(Lederle)实验室里从金色链霉菌中分离出来的;普菲泽尔公司的科学家们则从龟裂链霉菌中提取了土霉素,在此以前,他们曾检验过116,000个不同的土壤试样。到50年代人们发现这两种物质都是四环素的衍生物。而四环素则是从一些天然物质中分离出来的。这些化合物能有效地控制各种各样的细菌疾病、病毒性疾病和由立克次氏体引起的疾病。因此他们被称为广谱抗菌素。
抗菌素亦可用于动物饲养和用于禽肉、鱼肉和肉类的保鲜。在牲畜饲养中,由于抗菌素控制了进入牲畜消化道内的细菌,因而明显地提高了食物的利用率。
第二次世界大战之前,磺胺是唯一可以得到的有效的抗细菌药物。二次大战期间及战后,抗菌素研究在降低人、畜的致病率方面有着重大作用。
1945-1965年期间,青霉素开始大量使用,同时发现了头孢菌素。当时四环素、氯霉素、红霉素和氨基葡糖苷被用于治疗各种传染病。除了用发酵得到各种抗生素外,还开发了人工合成抗菌素,例如萘啶酮酸和硝基呋喃。在过去20年里,人们已作出了重大努力来改进临床使用的抗菌素的作用范围、效能和安全性。这包括发现新的发酵产物、欠佳的天然产物的化学修饰(半合成),以及用人工合成法引进新的结构类型等。在更加新的半合成青霉素中,有的药物不仅能抗普通的革兰氏阴性菌,而且能抗假单孢菌。这些假单孢菌在医院环境中正越来越成问题。早期的头孢菌素已经成功地被修饰成为具有显著广谱性、高效能和更加安全的新药物。
抗菌素研究的许多精力都曾用在对付产生抗药性的问题上,特别是医院环境里的抗药性。不幸的是,由于长期使用此药,细菌能产生抗药性,从而使抗菌素无效。例如,某些病菌能产生使抗菌素失活的酶。目前在设计合成破坏这类病菌酶的抑制剂方面已有了一些进展。另外一类细菌能通过阻止抗菌素进入菌体细胞而对其产生抗药性。当然,在这方面通过半合成衍生物及新药的发现,也取得了一些进展。
五、类固醇类药物(454-456)
有一大类重要的天然化合物是由四环结构的甾族化合物衍生出来的。这类化合物名为类固醇,它们存在于一切动植物体内。动物体内含量最多的类固醇是胆固醇C27H460。人体能合成胆固醇,也很容易通过肠壁吸收食物中的胆固醇。胆固醇和生成胆石有关,它还可使动脉硬化。
胆固醇的生化更迭和降解产生许多在人体生物化学中非常重要的类固醇。
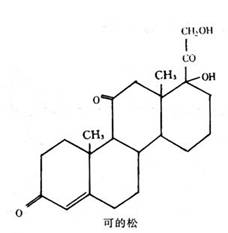
可的松和促肾上腺皮质激素(ACTH)是在1949年医药工业所生产的一种治疗风湿病和风湿性关节炎的药物。梅奥医学研究所的E・C・肯德尔(Edward C.Kendall)首次从动物的肾上腺皮质中把可的松分离出来。有关皮质激素的主要研究工作是在30年代里,在瑞士的梅奥医学研究所和雷依兹斯丁实验室里进行的。也在研究所里工作的P・S・亨奇(Philip.S.Hench)在几个关节炎和风湿病例中证实了该药物的药效。制作1克可的松需要180,000只绵羊肾上腺。这些可的松的化学成分马上被化验出来了,伍德沃德所领导的小组证实了该化合物的化学结构,并在1953年继续进行合成这种化合物的工作。由于它是一种甾族类的化合物,用于合成该化合物的原料容易得到,因此虽然其化学结构较复杂,但是进行工业合成生产还是可能的。
|
|
ACTH这种从猪脑垂体中提取的物质被证实是一种多肽。美国氰氨公司、加里福尼亚大学和匹茨堡大学的各研究小组一直在积极地从事着它的化学结构的研究。1960年在七年研究之后,由K・霍夫曼(Ⅺalas Hofmann)领导的匹茨堡小组合成了一种具备ACTH所具有的全部生物活性的多肽。
美籍华裔化学家李卓浩成功地确定了促肾上腺皮质激素(ACTH)的氨基酸数量(39个)和序列,ACTH刺激肾上腺因此控制像可的松这样的激素在体内的水平。他们发现主要的生物活性是由l-24的氨基酸所执行的;序列25-33氨基酸似乎表明该激素的人类血统,因为该段氨基酸随人种不同而变异,该序列中的其余成员(24-39)的功能还不清楚。
各药物实验室和大学所作的大量的研究使各种具有更高效的化合物设计工作稳步发展。特别是对甾族类化合物的研究对于改进可的松分子这方面起了不小的作用。氢化可的松是其中的一种,但它有许多不良副作用。其他有关化合物随着不饱和状态不断增加,和在某些位置上增添氟基,增添羟基或增添甲基,则药物的疗效更好。
性激素在结构上和胆固醇及可的松有关。女性激素(孕甾酮)和男性激素(睾丸甾酮)仅有不大的差别。
|
|
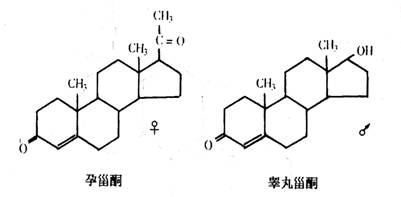
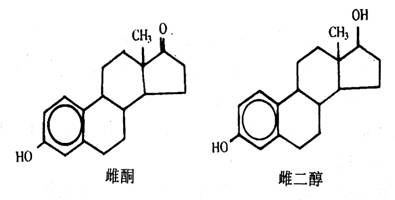
其他女性激素有雌二醇和雌酮,它们又叫雌激素。雌激素和前面讨论的其他类固醇的区别在于它们都有一个芳香族A环。
|
|
六、控制和诱发生育力的药物(456-458)
20世纪60年代医药界的一项革命性进展就是在全世界范围普遍采用女用口服避孕药。在美国现在有一千多万妇女使用节育药丸。女用口服避孕药的基本特点是它们具有模拟怀孕时产生的激素作用的化学能力,因而它们能阻止排卵。妇女怀孕开始后因激素改变即停止排卵,卵巢不再产生卵细胞。采用多种类固醇,其中有些可以口服,能产生相同的效果,虽然对它们的作用机制和长期效果知道得还不很详细。
女用口服避孕药的有效成分是孕甾酮和雌激素,或是它们的衍生物。
当我们发现了位于大脑深部的下丘脑和脑下垂体的作用时,对人类生育周期的认识随之有了很大的进展。下丘脑和脑垂体产生激素和神经传感质,用来控制生育周期,同时还控制体内其它激素的释放。这样,机体就控制了从卵巢释放卵子到乳汁分泌的一系列反应。当化学家们确定了这些激素的分子结构后,我们就有可能控制人的生育能力。
口服避孕药,已对世界人口的控制产生了巨大的影响。它们包括雌激素和孕酮两类化合物及其合成的类似物。不幸的是,在最初使用时,它有许多副作用。例如血栓、偏头痛、中风和心肌梗塞等都与它们的使用有关。过去几年里,临床上已注意减少雌激素和孕酮的剂量,同时选配好两者之间的比例,使口服避孕药的副作用大大地减少。
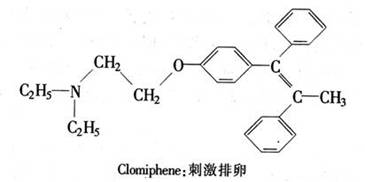
化学方法也被用来促进生育。 Clomiphene抑制下丘脑和脑下垂体中的雌激素受体。在妇女的生育周期里,选定合适时间,服用这种激素拮抗剂,就能干扰雌激素对下丘脑和脑垂体的正常反馈作用。这种干扰造成下丘脑和脑垂体的激素分泌高潮,常常引起排卵和受孕。
促性腺释放激素(GnRH)是由下丘脑分泌的。它刺激脑下垂体释放许多与生育有关的激素。含1O个氨基酸的多肽GnRH的许多类似物已经化学合成,并进行了试验。因为它们能产生某些副作用,所以人们对把这些类似物用于避孕的热情不高。尽管如此,人们仍然对它们抱有兴趣,并注意到它们对某些肿瘤的治疗效用。在先天性GnRH缺陷患者(很少见)的治疗中,这种GnRH类似物具有极佳疗效。在治疗过程中,临床医生把GnRH类似物放在小巧玲珑的泵内,以便患者携带,这样药物就能以搏动方式模拟脑下垂体有节奏地释放出来。使用过这种药物后,根本没有经历过青春期的一些二十多岁的患者,就能够通过青春期所有连续的内分泌阶段,然后成功地达到生育。给人印象深刻的药物设计和先进的投药系统的结合,是生殖研究领域中将来发展的一种前景。
|
|
另外,还有一些新的主要研究方向,在治疗上也取得了重要进展。几个实验室的实验结果表明,我们很快就会知道抑制素的分子结构。它是调节精子产生的关键性激素。通过合成这种结构的类似物,就有可能使药物化学家研制出用于男性的避孕药。可以预料,这样的化学药物,与女性使用的口服避孕药相比较,其副作用肯定会小得多。
七、维生素(458-461)
在整个人类历史上,缺乏维生素一直是死亡的重要原因。在18世纪,人们发现少量的柑橘果实可以防止长途航海中的坏血病。这是因为柑橘果实提供了维生素C。1912年,科学家把这种人体必需的“食物附加因子"命名为维生素。从那以后,许多维生素相继被分离鉴定。虽然维生素本身不是酶,但它对多种酶的作用是必需的。因此,它们被称为“辅酶”或“辅助因子”。下面介绍这方面的几项进展和新发现。
|
|
维生素B12是为防止恶性贫血和日常食物中所必需的组分。它的分离和鉴定在1948年就已有报导。1956年,科学家们用X射线晶体衍射和化学研究法,测定了它们的分子结构,发现它是现有维生素中最复杂的。1976年,Bl2的人工合成是有机化学的里程碑。在认识维生素B12辅酶的功能和作用机理方面,也有了很大进展。
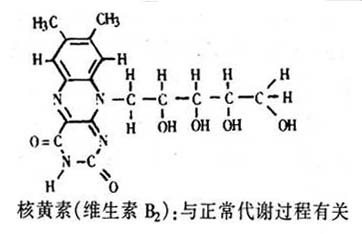
此外,人们对核黄素的认识也有了相当大的进展。核黄素,即维生素B2,是黄素的一个例子。各种形式的黄素是正常代谢过程中所必需的各种氧化还原系统的辅酶。现已知道有一百多种黄素蛋白。有趣的是近来发现一种修饰的黄素是产生甲烷的细菌中的一种辅酶,它在开发甲烷作为能源的研究中,可能是有意义的。
|
|
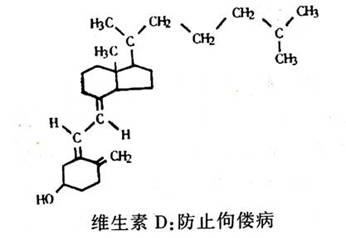
人们很早就知道维生素D是防止佝偻病所必需的。儿童缺少维生素D会使骨骼发育不良。利用先进的化学和光谱技术,现在已证明维生素D实际上是一种激素前体。在体内它被代谢成一种活性很强的二羟基衍生物,调节着吸收食物中的钙和在肾脏中钙的再吸收,以及钙在骨骼中的代谢。现在,人们还不了解维生素D激素如何完成它的功能,研究工作还在进行中。科学家们已经人工合成了维生素D,并证明它对许多骨骼疾病是很有效的。目前正在进行试验,以便正确估计它对治疗骨质疏松症的效用。这种化合物是极好的研究材料,随着研究工作的深入,无疑还会发现维生素D的新功能。
|
|
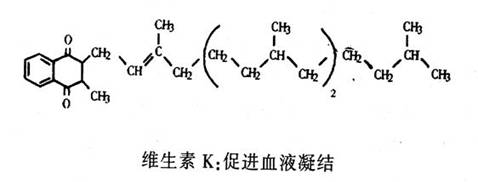
另一种结构已知的是维生素K。在产生帮助血液凝结的三四种蛋白质的过程中,它是必需的辅酶。我们仍需要弄清楚维生素K是如何起作用的,对其结构的了解是达到此目的的关键步骤。
|
|
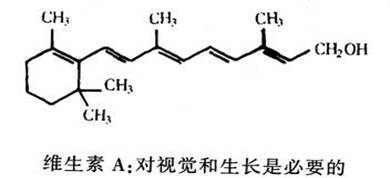
在相当长一段时间内,我们就知道来自维生素A的一种化合物是眼睛感光所必需的。然而,现在人们也认识到维生素A在高等动物的生长中也起着基本的作用。它在骨骼发育、精子发生及胎盘发育中都很重要。维生素A必须先转化成几种有关的化合物,才能发挥上述功能。目前,在阐明这些化学变化方面已取得了重要进展。例如,为了在上皮组织中发挥功能,似乎它必须先转变成视网膜酸。其中有些酸和人工合成的类似物,在治疗皮肤疾病,如痤疮方面是很有效的。另一重要进展是发现维生素A能够阻碍某些化学致癌作用。
维生素E,亦称“生育酚”。属脂溶性维生素。极易被氧化从而保护其他物质不被氧化,故具有抗氧化作用。是某些动物维持生殖机能的重要因素,缺少它会造成不育、流产等,对人类生殖机能的重要作用尚不甚明确,但在临床上常用于防止流产和不育等症。近年来有多家报刊报导,由于维生素E具有防止心脏病、减缓老化过程、减少人患白内障及某些癌症的作用,这种维生素已被人们称之为90年代的“奇迹丸”。在美国,1996年仅维生素E的销售额就达3亿美元,占所有维生素销售额的1/3。科学家认为,维生素E最大的好处在于它是一种抗氧化剂,可以控制对细胞的破坏,可以抗心血管疾病的作用,可以停止坏胆固醇的氧化。
与其他维生素不同的是,普通食品中所包含的维生素E的含量不够。研究指出,为有助于心脏健康,需要每日服用400个国际单位的维生素E。各种油,富含油果核拥有大量维生素E,因此日常应吃这类食物。应当指出,吃多了也有坏处。
八、心血管病类药物(461-464)
据卫生部门统计,中国共有心血管病人达1亿人,每年因此而死亡的人数超过了200万。心血管病(包括高血压、冠心病、脑中风)已成为危害人们生命、威胁人们健康的一大瘟疫。近年来,心血管疾病在美国已成为主要死亡原因。因此,高血压和高胆固醇也成了热门的研究课题。
高血压病
美国在1968-1978年间,冠心病的死亡率下降了20.7%,中度和严重高血压病防治上的改进,对这种下降起了促进作用。
最早的治疗高血压药物有严重的副作用。因此,只有当血压高到危及生命的时候才使用它们。现在有数种抗高血压药被广泛地用于防治中轻度高血压病,它们几乎无任何副作用。
肾上腺素是刺激神经自主作用的激素,包括使心脏不停跳动的自主作用。其分泌受类肾上腺神经系统的调节。虽然人们对引起继发性高血压的原因还不清楚,但类肾上腺神经系统和它的化学信息(去甲肾上腺素)在心脏功能及血压调节方面的作用早已得到公认。多年来,化学家们为临床医生提供了许多影响肾上腺系统的抗高血压制剂。Α-甲基二羟基苯丙氨酸是治疗高血压的最有效药物。
|
|
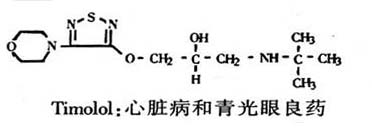
它以类肾上腺素受体的形式作用于中枢神经系统。鉴于去甲肾上腺素能作用于几种不同受体亚型,因此,我们就能够根据不同机理设计降血压药物。Timolo1和心得安是广泛使用的抑制去甲肾上腺素的两种药物。它们能有效地治疗某些心脏病,减少心脏病的复发和死亡的危险。Timolol还是治疗青光眼的药物。
其它两种类型的抗高血压药物是:在咽痛和中风治疗中也很重要的钙通道阻断剂和被称为血管紧张素转换酶的抑制剂。例如,巯基甲基氧丙基左旋脯氨酸和enalapril,它们对治疗心力衰竭也很有价值。
最近,化学家与生物学家一起发现、鉴定并合成了一组在心脏里释放出的多肽,命名为前心房促尿钠排泄因子。目前正在研究它们的生物学性质,以便确定对发展新的治疗剂的潜力。我们知道这些化合物具有利尿、舒张血管及降低血压的作用。
动脉粥样硬化
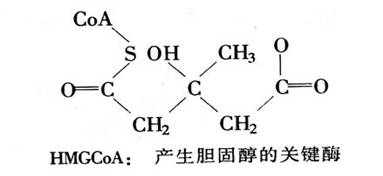
心血管病的第二个主要危险因子是血液中胆固醇过多,即高胆固醇。多年来,人们正在精心探索安全有效的药物。这些药物将通过阻止胆固醇的合成,或促进它的代谢,把血液中胆固醇水平降到正常范围。 3-羟基-3-甲基戊二酸单酰辅酶A(HMGCoA)还原酶,在肝脏中对胆固醇的形成起着重要作用。由于现在有了一种新的能作用于MGCoA的酶抑制剂,这就使高效治疗高胆固醇有了希望。
|
|
心力衰竭
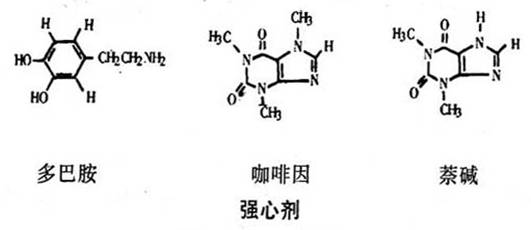
毛地黄尽管有严重的副作用,但两个世纪以来,它一直是治疗心脏衰竭的主要药物。研究者们正在寻找能改善衰竭心肌机能、而毒性又小的药物。现在对高水平CAMP(cyclic adenosine monophosphate)剌激心脏收缩的研究比较充分。细胞中CAMP的水平可通过prenalterol、多巴胺、多巴酚丁胺的作用直接提高,也可通过咖啡因或茶碱间接提高。这类药物能抑制使CAMP失活的磷酸二酯酶。
自70年代开始的10年中,在对出血性心肌衰竭治疗中,除使用传统药物毛地黄和利尿剂外,常辅加一些其它新药。有时候则完全用新药,而不用毛地黄和利尿剂。这些新药物对心脏没有直接作用,但它可通过血管扩张使心跳加强。在80年代里,这些新的血管舒张药(如上面提到的巯基氧丙基左旋脯氨酸和enalapri1)对出血性心力衰竭的处治,可能已产生明显作用。
|
|
心律失常
另一类常见心脏病是心律失常。当今两种广泛使用的抗心律失常药物是奎尼丁和毛地黄。它们的起源可追溯到200年以前。从18世纪以来,这类药物就已用于治疗具有潜在危险的心律失常症。
|
|

现在我们在研究这类化学药物作用机理方面,已取得了许多进展。心脏跳动的节律是受Na+――Ca2+泵的电信号控制的。现已发现奎尼丁、普鲁卡因酰胺和利多卡因可使钠通道失活;维拉帕米(verapamil)能抑制钙通道;心得安、噻吗咯尔能抑制交感神经的活动;而胺碘达隆(Aminodarone)则可延长神经冲动。这些药物构成了目前治疗心律失常的基础,并指出了合理治疗的途径。
九、影响中枢神经系统(CNS)的药物(464-466)
用于直接护理精神病人的费用占全美国医疗卫生总费用的15%。每年大约有2.5%的美国人接受精神病或情绪异常的治疗。抗抑郁药和镇静药能使那些精神紊乱的人,过正常人一样的生活。
早期的治疗精神病的药物都是通过临床症状观察和粗糙的试验发现的。因为没有可靠的理论依据,因此,就必然导致化学家在合成更理想的治疗药物方面的工作进展缓慢。最近,化学家与神经生物学家开始合作,着手探讨这些药物治疗效果的生化机理。结果,对精神病、抑郁症和忧虑症可产生疗效的各种方法正在不断出现。
|
|
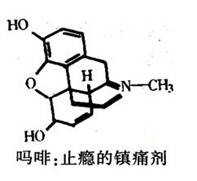
在作用于中枢神经系统的镇痛药物中,最重要的那些都来自鸦片。广泛使用的传统镇痛药吗啡,正在被人工合成的新药所取代。这种新药服用后不会上瘾,也没有什么副作用。此外,现在已有了治疗海洛因、鸦片和吗啡上瘾的药物。 20世纪70年代,从脑组织分离出了两种在功能上类似吗啡的多肽,即脑啡肽,并进行了化学鉴定和人工合成。这一发现,对研究CNS具有深远的影响。
Try-Gly-Phe-1eu
亮氨酸.脑啡肽链:一种新的镇痛药
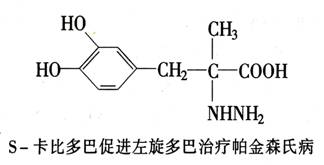
中枢神经系统生物化学治疗的典型例子是帕金森氏病。该病的症状是抽筋和瘫痪。这是由于多巴胺短缺而引起的。现已知道,该病可用左旋多巴来治疗。该药能进入脑组织,并在那里被多巴脱羧酶转变为多巴胺。化学家把左旋多巴与卡比多巴合并使用,导致该研究取得了进一步的进展。卡比多巴能防止左旋多巴在脑组织之外毫无用途地代谢,这样一来活性成分就只能在需要的地方,即脑组织中形成,因而副作用大大减小。
|
|
80年代以来,我们对哺乳动物中枢神经系统中发生化学信号过程的认识有了明显的进步。 20年前,我们只知道有8-9种单胺和氨基酸化合物似乎是神经传感质,而现在却可以列举出40多种小分子肽。每种多肽都可能有信息功能。通过化学和生物学联合进行研究,在治疗上出现重要进展的机会是巨大的。
十、癌症的研究(466-469)
癌症在美国造成死亡的人数仅次于心血管病,而且对美国四分之一人口构成威胁。癌症的特点是细胞在体内无限制的增长。令人宽慰的是,癌症研究已进入了一个富有成果的时代。现在对癌的起源,即癌的发生的认识和癌症的化学治疗这两个方面都已取得了新的进展。
癌的发生
30年代,科学家们发现某些有机化合物能在实验动物体内致癌。后来又找到许多不同的化合物,在足够剂量条件下,都能在小鼠、大鼠和其它哺乳动物的组织中诱发癌症。今天,在环境中某些天然的或人工合成的化学物质,被怀疑能诱发人的癌症。因此,对检测这些因素和研究其作用机制的兴趣越来越浓了。
1965年以前,就已确认了化学致癌剂及其致癌作用的几个显著特性。并证明了几种不同的化学致癌剂在活体内以共价结合到细胞的大分子(蛋白质、RNA和DNA)上,这种结合是与致癌过程有关的,这些发现为进一步的研究奠定了基础。
大多数已知的化学致癌剂,实际上是“原致癌剂”。也就是说,它们必须在人体内经过激活才能成为最终的致癌剂。例如,苯并芘是一种原致癌物,它参与一系列酶促反应,最终产生与DNA结合的致癌物。这种结合了致癌物的DNA分子称为DNA加成产物。
|
|
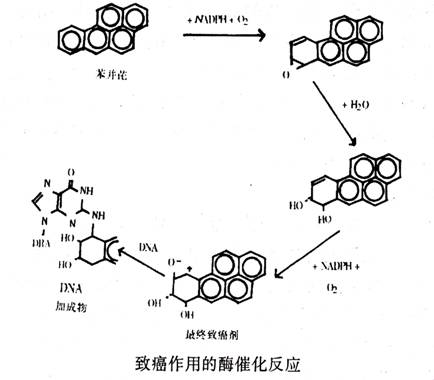
正是由于这种最终致癌物与细胞内的核酸和蛋白质的结合,才打乱它们在细胞正常生长中的功能。科学家们已经对转化原致癌物的主要酶系统进行了研究和鉴定。人们对致癌物――DNA加成产物形成的化学基础已很清楚。但对这类加成产物如何使动物体致癌却不大了解。现在已知当致癌物在体内经化学加工时,其最终产物会使细菌和动物细胞中的DNA发生变化(突变效应)。突变因子与致癌物之间存在着一定的关系。当一种化合物被证明是突变因子时,它往往也是致癌物。这种估计可在实验室内用埃姆斯实验常规进行。该实验用的材料是沙门氏杆菌(Salmonells)的一种特殊菌株培养物。但并不是所有的突变因子都是致癌物。在我们普通的饮食中就存在有大量的天然突变因子。
当细胞变成恶性癌细胞时,其生长异常,此时即被认为对生命造成威胁。也许近来在癌症研究中最引人注目的进展,是发现了在正常细胞中有与导致细胞恶化有密切关系的一些基因。值得注意是:这类基因与使正常细胞转为恶性细胞的病毒的基因(癌基因)相似或相同。有机化学家能够测定:①正常基因和致癌基因的核苷酸序列;②由这些基因编码的蛋白质的氨基酸序列。当膀胱、结肠和肺细胞的一个基因中的一个核苷酸被改变时,就能使基因产物中的一种特定氨基酸被另一氨基酸所取代,结果使得正常细胞成为恶性细胞。这一发现的重要意义是:我们在分子水平上认识了某些转化作用中正常细胞与癌细胞的蛋白质之间的差异。这种认识使我们能更接近于合理地研究新的治疗方法。
化学疗法
用于治疗癌症的化合物原先是从自然界分离的或人工合成的有毒物质。药物化学家的任务是设计和合成疗效有所改进的新药。过去25年里,科学家们已从微生物中分离了许多临床上重要的抗肿瘤药物,并测定了它们的化学结构。在多种这类化合物中,有可能制备出毒性和副作用大大减少的半合成产物。某些抗生素是通过插入到恶性细胞的DNA双螺旋里而与DNA相互作用的。这种机制为设计正在作临床试验的新合成药物提供了模式。
第一个人工合成的抗癌药定名为氮芥气,它是使DNA烷基化而起作用。此后,又合成了许多类似的化合物,出现了更有效的药物,如环磷酰胺。它们具有更好的选择性,即只作用于患变细胞的DNA。另外一类广泛使用的抗癌药物是“抗代谢物”,这是模拟干扰代谢过程的天然物质而合成的。还有一些化合物带有高电子亲和性,如misonidazole,它能使癌细胞对放射治疗更加敏感。已证明大约有40种抗癌药在临床上是有用的。治疗上最显著的突破来自几种药物的综合疗法。在过去30年里,化学疗法的最重要进展是在治疗儿童癌症方面取得的。在几种肿瘤类型中,受尽折磨的患儿的成活率已从不到20%提高到60%以上。
据1997年2月报导集放疗和化疗的优点放射性抗癌药已在美国投入临床使用。
对于更有效而毒性较低的抗癌药物,特别是治疗生长缓慢的实体瘤、肺癌和脑癌的药物,仍然是一个迫切需要解决的问题。免疫学家和细胞生物学家正在揭示正常细胞和肿瘤细胞表面之间的差异,这些差异可能为药物设计提供新的方向。此外,在发现能促进宿主免疫反应的药物中,化学家们也将起着至关重要的作用。
综上所述,用大量例子说明我们已在分子水平上认识药物的化学作用。这些知识有助于我们在分子水平上治疗疾病,从而达到理想的疗效。因此,我们已经进入了一个能够合理而随意设计药物的时代。
十一、中国药物化学(469-471)
中国是中草药的故乡。1949年以前,中国化学家在中草药有效成分的研究方面已取得令人瞩目的成就,如麻黄素的药理作用、钩吻和汉防己生物碱等的分离及分析工作、雌性甾族激素的全合成等。在当时条件下,能取得这样的成绩,实属难能可贵。庄长恭、赵承嘏、曲呜龙、纪育沣就是中国第一代药物化学家。
1952年,原中国科学院有机化学研究所独立为中国科学院上海药物研究所,所长为赵承嘏,1966年赵承嘏病故由高怡生任所长,嵇汝运任副所长。中国医学科学院也成立药物研究所,有药物化学家黄量和梁晓天从事研究工作。北京医学院(现为北京医科大学)成立了药学系,由著名药物学家王序任系主任,1984年王序病故后由王夔任系主任。从50年代到60年代前半期,中国的药物化学得到了蓬勃发展。在为改善人民卫生健康而向科学进军的征途中,中国化学家如汪猷、邢其毅、朱子清、黄耀曾等也曾参加药物化学研究。
赵承嘏等很早就从麻黄中分离出左旋麻黄素及其副碱左旋甲麻黄素;庄长恭等从汉防己分离出防己诺林碱,证明其为脱甲基倒地拱碱;以后,邢其毅研究了防己诺林碱分子中酚羟基的位置在C7而不是在C6;高怡生、赵承嘏从木防己分离得木防己甲、乙两素,乙素经甲基化即得甲素;赵承嘏、朱子清在1949年以前就从事浙贝母中生物碱的研究,确定了贝母素甲与乙为仲醇和酮的关系;1954年朱子清等对贝母素甲、乙及酉贝素的结构进行了研究,证明它们是变型甾族生物碱,这一贝母生物碱的基本骨架是中国有机化学家提出来的,并已为国际化学界所接受。
在抗疟药物方面,中国科学工作者曾调查分析出多种抗疟中草药,其中有常山和青蒿。常山含有常山碱,其中γ-异构体有显著抗疟作用、效力超过奎宁148倍,但毒性也较大。
由于萝芙木碱具有降低血压的功能,50年代中国化学家和医药工作者曾在国产中药中广为搜寻,并对其化学性质、结构及药理作用取得有意义的结果。
60年代,在中国发现喜树中的喜树碱也具有抗癌功能,中国科学家对这类植物进行了普查和系统分离鉴定,有的已合成成功。中国科学家首先完成了对四种能抗癌的尖三杉酯的半合成工作。
钩吻是治疗癌症的中药,30年代至40年代初,赵承嘏、朱子清就曾从国产钩吻中提取它的有效成分,证明其为生物碱;1961年朱子清、刘铸晋等用化学降解和光谱分析测定了钩吻的部分结构;最近,刘铸晋等进一步用核磁共振阐明了它的完整结构,属于新类型吲哚生物碱。
中国化学家曾在国内植物中分离得藤黄酸、别藤黄酸、冬凌草乙素、新疆紫草等抗癌物质。
中国化学家对抗瘤药物美登木素也进行了植物普查和分离鉴定,证明它是一个结构复杂的具有8个不对称碳原子的大环酰胺化合物;高怡生等从事着艰巨的全合成工作,已合成了大环化合物的两个半环分子,特别是内酯化这步合成工作是很出色的。
中国在30年代开始从事男性激素的研究,庄长恭曾报导雌性激素类似物去甲脱氢雌马甾醇的全合成。1949年后,中国甾族化学得到真正的发展。黄鸣龙等合成了肾上腺皮质激素、女性激素及甾族口服避孕药,并投入了生产。为了适应计划生育的需要,中国有机化学家和医药化学家合作,开展了甾族口服避孕药物的合成与动物临床试验。黄鸣龙等通过七步反应合成可的松已用于工业生产。
在抗生素研究工作中,桔霉素是较早的一例。在化学方面,汪猷等曾进行了一系列结构与合成的研究。
1953-1955年,中国科学院和中国医学科学院的有关研究所、化学工业部医药工业研究部门曾同高等院校通力合作,对金霉素、土霉素、链霉素等从菌种、发酵、分离、化学性质与鉴定直到中间试验进行了一系列研究,后来又对四环素和青霉素的生物合成进行了研究。
黄耀曾对金霉素进行了化学降解研究;朱秀昌等合成了性质优良的阳离子交换树脂,可以从发酵液中分离链霉素类碱性抗生物质并用于生产;汪猷曾证明Wolfrom提出的链霉素结构有部分错误。
氯霉素的研究主要反映在全合成的工业生产方法上。邢其毅和戴乾圜应用曼尼希反应合成ω-甲胺基对硝基苯丙酮盐酸盐和利用普林斯反应合成中间体,为制造氯霉素提供了新的合成方法。
创新霉素完全是中国科学家分离,测定结构并合成的一种新抗生素。
十二、新药设计的现状与趋势(471-474)
1.国内现状及要求
建国以来,我国已建立了一个比较完整的医药工业体系,我国生产的原料药品种已达千种以上,年产量也超过10万吨,基本上保证了人民用药的需要,但与世界先进水平相比差距仍相当大。目前我国生产的绝大多数药品均系仿制国外研制的新药,而我国自行设计创制的新药,据统计先后曾有100余种,但能大量生产供应市场的一线药物很少,迄今还没有一个新药能在国外注册,进入国际市场。1987年我国出口的药品主要是一些专利过期的仿制品,虽达2万多吨,但仅换取了3亿多美元,而同年SKF药厂的一个创新药品甲氰咪呱的年销售额就达7亿美元。因此若没有自己的创新药品是不可能参与国际竞争的。面对目前的形势,必须立刻加强新药的研究与开发。即使如此,其研究成果(新药上市)亦将在2000年以后,这是因为一个新药的开发周期(自研究至生产)平均约需10-15年。
2.药物设计的现状与趋势
药物化学的任务是整个新药研究开发的前期,即设计并合成有效的化合物。传统的方法是合成大量的化合物进行药效筛选,命中率约为万分之一,研究的周期长,因此耗资甚大。但由于成功的新药利润高,因而一些制药企业都把新药开发作为其生命线。为了提高成功的可能性,创制新药通常多选用具有新型结构并有确切药理作用的天然产物或已知的合成药物为先导物,通过药物设计进行化学结构的修饰、简化,使之更能适应于疾病的治疗。随着许多重要生命过程的调节机理以及病理变化因素,特别是体内神经介质、酶、受体等在生命过程中的调节作用被逐步阐明,调节体内某些过程用以控制疾病的发展或纠正病理变化已经成为新药研究追求的主要方向。近年来国际上在新药设计上的一些趋势分别是:
(1)酶抑制剂的研究。许多药物能与酶结合,改变了酶的特性,使之不能再起类似的催化,从而干扰了有关的生化反应,产生了药物的效应。例如毒扁豆碱或新斯的明用以治疗重症肌无力,青霉素或头孢菌素类抗生素干扰了细菌的羧肽酶和转肽酶,使细菌不能合成细胞壁,从而导致细菌的死亡等。近年来揭示了大量酶反应的历程细节,也阐明了一些酶的三维结构。据此有可能设计具有一定目标的新药物。其中受人注目的有血管紧张素转化酶(ACE)抑制剂及肾素抑制剂和3-羟基-3-甲基-戊二辅酶A(HMG-COA)还原酶抑制剂等。
生物体内有着众多的酶系,随着对一些在生理上至关重要的内源性物质功能的认识,各系统的酶的抑制剂将不断问世。
(2)受体拮抗剂。受体是一种特异性大分子。内源性的激素或神经递质,在极低浓度就能和有关受体相互作用,生成可逆性复合物,启动功能性变化,如开启离子通道或激活有关的酶,最终导致生理变化。药物作用在同一受体,若产生类似的生物效应则称为激活剂,若与受体结合,但并不随着产生生理作用,却阻碍了激素,递质等物质与受体结合则称拮抗剂或阻断剂。许多药物是受体的拮抗剂。大多数受体是嵌入于细胞膜的脂蛋白或糖蛋白,其结构较酶更为复杂,除了少数受体的结构已经深入研究外,大多数受体的结构还不甚明了。往往只能通过一系列激动剂和拮抗剂,以间接推测受体和药物结合的方式。由于受体蛋白长长的肽链很易缠卷,常有一个或几个低自由能的构象存在,互变的能障也低,因此可以存在几种不同的亚型。例如肾上腺素的受体有α-型和β-型,而α-型又可分为α-1及α-2,β-型也可分为β-1及β-2,多巴胺受体可区分为DAl和DA2型。药物结构有所改变时,与不同构象的受体亚型的亲和力可发生变化,若所成复合物能量愈低则愈稳定。药物结合在不同受体亚型可产生不同效应,合理性药物设计在于寻找特异性强,专一地作用于某一亚型的药物。
如上所述,随着人们对各种受体结构,功能的逐步了解,今后有可能设计各种具有不同专一性功能的药物。
(3)已知药物的结构改造。在新药发现中,药物化学的主要作用有二:一是发现先导化合物,该过程可以是广筛的结果,也可是合理设计的结果;二是根据先导化合物,进一步优化,即化学结构的修饰、简化,获得实用的药物。最近几年,国际上每年约创制新药60个左右,而真正导向研究而获得全新结构的并不多,大部分均属于现有药物的结构改造。当一个有效药物问世后,往往就其基本结构作各种改变,从而进一步提高疗效,改进副作用。此外亦可避开其专利权而开发具有相似功效的新化合物。如巯甲脯氨酸(Captopril)的成功,导致了ACE抑制剂研究的高潮。人们发现Captopril分子中的巯基与其副作用――过敏性皮疹、味觉障碍、半衰期短――有关。由此Merck药厂开发了新药苯酯丙脯酸(Enalapril),其疗效优于Captopril。利用计算机辅助设计新药特别受到重视,如氟哌酸(Norfloxacin)的问世,导致了氟化喹酮酸抗菌药物研究的高潮,据统计1991年喹酮酸类抗菌药物的年销售额超过13亿美元,是抗菌药物中年增长率最高的一类。有人将氟代喹酸类药物的开发称之为抗菌药物的第四代革命。(本章完)