测试与标准
IP-HPLC法测定织物中致癌染料含量qt-081207-1
丁友超, 蔡建和, 杨锦富, 曹锡忠, 杨安南 (江苏出入境检验检疫局,江苏南京210001)
收稿日期:
2008-06-19
基金项目:“十一五”国家科技支撑计划重点项目,出入境检验检疫安全关键技术研究(项目编号:2006BAK10B03)。
作者简介:丁友超(1977-),男,工程师,博士研究生,主要从事生态纺织品研究。
原载:印染2008/15;38-41
网上来稿: zhanyizhen
08-8-7
【摘要】 采用不同试剂蒸汽作为剥色剂剥离织物纤维上的染料,用离子对反相液相色谱法( IP2HPLC)对不同织物上的9只禁用染料同时进行测定。用吡啶/水(4/3,v/v) 、吡啶/甲酸/水(20/5/75,v/v)和氯苯可分别完全剥离天然纤维上的直接染料和酸性染料,腈纶纤维上的碱性染料和涤纶纤维上的分散染料。以磷酸二氢四丁基铵作为阳离子对试剂,以增加阴离子染料的保留,在反相色谱柱上通过乙腈洗脱,可完全分离这9只染料,再用二极管阵列检测器在最大吸收波长检测染料含量。该方法中, 9只禁用染料的定量限为1.0~10.0mg/kg,线性相关系数都大于0.995,回收率为92.3%~106.8%,批间的相对标准偏差(RSD)均小于10.0%。
【关键词】测试; 含量; 致癌染料; 液相色谱法
【中图分类号】TS197 文献标识码: C 文章编号: 1000-4017(2008)15-0038-04
0 前言
C.I.直接蓝6、C.I.直接红28、C.I.直接黑38、C.I.酸性红26、C.I.碱性红9、C.I.碱性紫14、C.I.分散蓝1、C.I.分散橙11和C.I.分散黄3,是欧盟指令1999 /43
/EC禁止销售和使用的9 只致癌染料。欧盟委员会的纺织品生态标签Eco2label(欧盟2002/371/EC决议) ,也要求被授予生态标签的纺织品不得使用这9只致癌染料;而国际环保纺织协会颁布的生态纺织品标准Oeko2Tex Standard 100,则要求纺织品中每只有害染料的检测限量为50mg/kg[1] ,对纺织品中致癌染料的检测提出定量要求。
GB/T
20382-2006《纺织品致癌染料的测定》[2]用超声波辅助甲醇方法提取织物纤维上的这9只致癌染料,并在高效液相色谱仪(HPLC2DAD)上检测。该方法解决了纺织品中致癌染料难以检测的难题,且超声波辅助甲醇的提取方法,可一次提取不同类型纤维上的多种染料,方法简便、快捷。但实践表明,该方法只能提取纤维上的部分染料,回收率较低,不能满足Oeko-Tex Standard 100的定量检测要求。
依据染料对纤维的染色机理,本试验选用不同剥色剂,对纺织品中四类致癌染料完全提取,用离子对高效液相色谱法(IP-HPLC)进行染料分离和测定,方法灵敏、准确,技术指标符合定量分析法要求,且满足生态纺织品的检测要求。
1 试验
1. 1 仪器与试剂
仪器 Agilent 1200高效液相色谱仪(美国,配有二极管阵列检测器);可控温超声波仪(输出功率420W,频率40kHz,70℃时控温精度为±2℃,美国Branson公司);剥色装置(自制,见图1);可控真空旋转蒸发仪(德国BUCH
I公司); 0.45μm聚四氟乙烯薄膜过滤头(津腾公司)。
|
|
|
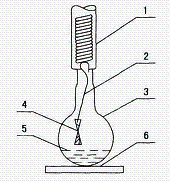
图1 剥色装置 |
|
1-冷凝水管; 2-金属钩; 3-烧瓶; 4-织物样品; 5-剥色剂; 6-加热平板 |
试剂 9只致癌染料有证标准物质(德国Dr.
Ehrenstorfer公司); 甲醇、乙腈(色谱纯, 德国Merck 公司); 5 M磷酸二氢四丁基铵(分析纯,美国Sigma公司);吡啶、甲酸、氯苯、二甲基甲酰胺、氯苯、盐酸等(均为分析纯);水(由MilliQ系统制备纯水)。
1. 2 参考样品制备
5块参考样品由上海染料研究所制备,分别用商品染料直接红28和直接黑38上染棉坯布,酸性红26上染毛坯布,碱性紫14上染腈纶坯布,分散黄3以不同的染色工艺上染涤纶坯布。
1. 3 样品前处理
取代表性样品剪成细条状,混匀。称取1.00
g试样(精确至0.01g) ,用无色纱线扎紧系于剥色装置的金属钩上,垂直置于20 mL沸腾的剥色剂上方30min。天然纤维(纤维素纤维和蛋白质纤维)织物用吡啶/水(4/3,v/v)
作为剥色剂, 腈纶纤维用吡啶/甲酸/水(20/5/75,v/v) 作为剥色剂,涤纶纤维用氯苯,采用IP-HPLC2DAD分析。
1. 4 仪器分析
色谱柱 Agilent XDB
C18反相色谱柱(100mm×2.1mm,5μm)
保护柱 C18柱(美国Phenomenex公司)柱温50℃
流动相 乙腈(A)和2.5
mmol/L 磷酸四丁基铵溶液(B)
梯度洗脱 35 min内A从20%线性增至100%
,并保持5 min。
流速 200μL/min
进样量 10μL
DAD扫描波长 190~700nm
定量波长 分散蓝1 在400nm, 分散橙11
在500nm,酸性红26、直接红28、碱性红9 和碱性紫14在540nm,直接蓝6、直接黑38和分散蓝1在600nm。
2 结果与讨论
2. 1 剥色剂的选择
本试验所研究的9只致癌染料分别属于直接、酸性、碱性和分散染料,化学性质差异较大,主要用于棉、毛、腈纶和涤纶等纤维的染色[2,3] ,因而需根据其染色机理,采用不同的试剂分别进行剥色处理。
剥色剂需满足如下条件:(1)仅能溶胀纤维,不溶解和分解纤维;(2)溶解染料,但不能与染料发生化学反应;(3)溶剂性能稳定、无色、不易燃。本试验以参考样品为试验对象,采用多种试剂剥离纤维上的染料,部分试验现象及结果见表1。
表1 各种试剂及前处理方法对纤维中致癌染料的提取情况
|
试剂 |
方法 |
棉 直接黑 38 |
棉 直接红 28 |
毛 酸性红 26 |
腈纶 碱性紫 14 |
涤纶 分散黄 3 |
|
甲醇 |
A |
1 |
1 |
1 |
1 |
1 |
|
甲醇 |
B |
1 |
1 |
1 |
1 |
1 |
|
甲酸 |
B |
1 |
1 |
1a |
1a |
1 |
|
盐酸/甲醇/水(2/1/1) |
C |
1 |
1a |
1 |
1a |
0 |
|
盐酸/甲醇/水(2/1/1) |
A |
1 |
1a |
1 |
1a |
1a |
|
吡啶/水(4/3) |
A |
1 |
2 |
2 |
1 |
0 |
|
吡啶/水(4/3) |
B |
2 |
2 |
2 |
1 |
0 |
|
吡啶/水(4/3) |
C |
2 |
2 |
2 |
1 |
0 |
|
吡啶/甲酸/水(20/5/75) |
C |
1 |
1 |
1 |
2 |
0 |
|
吡啶/甲酸/水(20/5/75) |
B |
1 |
1 |
1 |
2 |
0 |
|
二甲基甲酰胺 |
B |
2 |
1 |
0 |
0b |
2 |
|
吡啶/水(1/1) |
C |
2 |
2 |
1 |
1 |
0 |
|
吡啶/水(1/1) |
B |
2 |
2 |
1 |
1 |
0 |
|
氯苯 |
B |
0 |
0 |
0 |
0 |
2 |
注:前处理方法:A-70 ℃水浴, 420 W、40 kHz超声30 min;B-沸腾试剂剥色30 min; C-100 ℃水浴中静置30 min。0-无法提取染料,提取剂为无色; 1-提取纤维上部分染料; 2-完全提取染料,纤维没有颜色; a-发生化学反应,染料变色; b-纤维被溶解。
在国外考古研究中,酸性甲醇试剂(盐酸/甲醇/水,2/1/1,v/v)常用来提取天然纤维中的天然染料[4]。但对本试验研究的合成染料,酸性甲醇溶液与直接红28、碱性紫14和分散黄3都发生了变色反应。二甲基甲酰胺(DMF)虽可剥离棉纤维上的直接染料和涤纶纤维上的分散染料,但会溶解腈纶纤维,不适合混纺纤维的处理。试验表明,极性较强、能提供质子的吡啶水溶液(吡啶/水,4/3,v/v)[5],可完全提取天然纤维中的极性直接染料和酸性染料;极性较弱的氯苯[6],可完全提取化学纤维中弱极性的分散染料。对于极性碱性染料,需在吡啶水溶液中加入甲酸(吡啶/甲酸/水,70/5/25,v/v)[7]来置换腈纶上的阳离子染料。腈纶纤维上碱性染料的剥色原理如下:
腈纶-SO-3染料母体 + HCOOH = 腈纶―SO3H + HCOO-染料母体
2. 2 提取效率比较
通常,直接染料和酸性染料在常温下染色,碱性染料和分散染料在高温下染色[3]。因此,本试验在剥色装置中用剥色剂的蒸汽来提取纤维中的染料,这样既可利用蒸汽的高温提取染料,也可以避免提取液与纤维直接接触造成沾色。
分别在GB/T 20382-2006规定的超声波辅助条件下及在本试验研究的剥色条件下提取参考样品中的染料,二者的测定结果见表2。
由表2知,剥色法能完全提取纺织品中的染料,而超声波辅助甲醇法仅能提取纺织品中6.2%~56.3%的染料,提取率较低。因此,对于纺织品中含量较低的致癌染料,用超声波辅助法提取,可能会因提取量较低而难以检出,易导致结果的误判。
2. 3 色谱分离
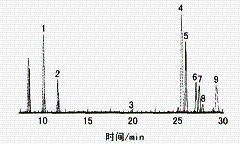
直接红28和直接黑38在水溶液中可电离成二价阴离子,直接蓝6可电离成四价阴离子,在C18柱上都难以保留。本试验用磷酸二氢四丁基铵作为阳离子对试剂,其中四丁基铵阳离子可与染料阴离子相结合,从而增加了阴离子染料(直接染料和酸性染料)的色谱保留[8]。图2(A)中,碱性红9和碱性紫14因电离生成极性阳离子,而在C18柱上最先被洗脱;接着是弱极性的分散蓝1;之后是含有磺酸根的阴离子染料,其因与阳离子对试剂作用而被洗脱;带四个磺酸根的直接蓝6,因与四丁基铵阳离子结合成极性更低的化合物,而在C18柱上保留最长。
|
|
|
|
(A) |
(B) |
|
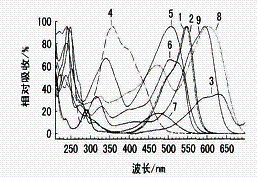
图2 35 mg/L的9只染料的液相分离色谱图(A)和光谱图(B) |
|
|
12碱性红9; 22碱性紫14; 32分散蓝1; 42酸性红26; 52直接红 28; 62分散黄3; 72分散橙11; 82直接黑38; 92直接蓝6 |
|
2. 4 光谱检测
图2 (B)为染料在200~700
nm的光谱图, 9只致癌染料在紫外和可见光区域内都有强的吸收峰。根据图2中的色谱保留时间和光谱图对染料进行定性分析。为了避免紫外区域的较强干扰,本文在可见光区域内靠近染料强吸收峰选取定量检测波长,见表3。
表2 用超声提取法( A)和剥色法( B)处理参考样品的平行测定结果(mg/kg)
|
序号 |
直接红28 |
直接黑38 |
酸性红26 |
碱性红9 |
碱性紫14 |
分散黄3 |
||||||
|
A |
B |
A |
B |
A |
B |
A |
B |
A |
B |
A |
B |
|
|
1 |
157 |
5870 |
523 |
1201 |
19.5 |
1769 |
52.7 |
498 |
817 |
3380 |
178 |
5623 |
|
2 |
162 |
5440 |
462 |
962 |
23.8 |
1520 |
75.4 |
402 |
828 |
3684 |
159 |
5857 |
|
3 |
109 |
6012 |
351 |
973 |
61.3 |
1632 |
89.7 |
411 |
668 |
3672 |
234 |
5430 |
|
4 |
115 |
5995 |
327 |
1121 |
35.8 |
1649 |
86.5 |
421 |
812 |
3441 |
249 |
5312 |
|
5 |
130 |
6123 |
498 |
1029 |
44.6 |
1902 |
105 |
387 |
836 |
3812 |
189 |
5388 |
|
6 |
157 |
5846 |
512 |
947 |
55.3 |
1886 |
64.5 |
454 |
731 |
3579 |
141 |
5044 |
|
7 |
146 |
6005 |
553 |
895 |
30.7 |
1796 |
93.4 |
381 |
676 |
3563 |
155 |
5116 |
|
8 |
152 |
6098 |
326 |
1128 |
31.5 |
1812 |
90.9 |
419 |
863 |
3521 |
236 |
5555 |
|
9 |
102 |
5769 |
543 |
1021 |
38.9 |
1782 |
78.9 |
406 |
851 |
3571 |
229 |
5063 |
|
10 |
176 |
6235 |
330 |
998 |
40.1 |
1761 |
79.6 |
412 |
677 |
3521 |
258 |
5314 |
|
平均值 |
141 |
5393 |
443 |
1028 |
38.2 |
1751 |
81.7 |
419 |
776 |
3574 |
203 |
5370 |
|
RSD/% |
17.8 |
3.8 |
21.9 |
9.2 |
34.0 |
6.8 |
18.4 |
8.2 |
10.22 |
3.5 |
21.3 |
4.9 |
表3 9只禁用染料的HPLC2DAD分析
|
分析物 |
tR /min |
检测波长nm |
线性方程( n = 6) |
线性范围(mg/L) |
相关系数γ2 |
定量限 (mg/kg) |
|
碱性红9 |
10.13 |
550 |
Y=-0.4953+12.7672X |
0.1~10 |
0.9998 |
1.0 |
|
碱性紫14 |
11.63 |
550 |
Y=-1.0614+66.9906X |
0.1~10 |
0.9999 |
1.0 |
|
分散蓝1 |
19.84 |
600 |
Y=2.3566+0.8109X |
1.0~100 |
0.9951 |
10.0 |
|
酸性红26 |
25.44 |
480 |
Y=11.8691+17.7837X |
0.5~50 |
0.9992 |
5.0 |
|
直接红28 |
25.91 |
480 |
Y=0.8504+15.8769X |
0.5~50 |
0.9999 |
5.0 |
|
分散黄3 |
27.05 |
360 |
Y=5.0647+23.6839X |
0.5~100 |
0.9998 |
5.0 |
|
分散橙11 |
27.42 |
480 |
Y=8.5015+21.1977X |
0.25~50 |
0.9995 |
2.5 |
|
直接黑38 |
27.76 |
600 |
Y=0.1696+5.1243X |
0.2~50 |
0.9999 |
2.0 |
|
直接蓝6 |
29.33 |
600 |
Y=-0.5509+11.9011X |
0.5~50 |
0.9999 |
5.0 |
2. 5 方法检测限及线性
以化合物峰面积(Y)对检测值(X,mg/kg)绘制标准曲线(表3)
,回归方程的线性较好(γ2>0.995)。以样品空白的10倍噪声计算定量限,
9只染料的定量限为1.0~5.0 mg/kg,均低于Oeko2Tex Standard 100要求的50mg/kg限量值。
2. 6 方法精密度与回收率
表2中,对参考样品进行10次平行测定,由超声波辅助甲醇法测定结果,其RSD 值均大于12.3% ,而剥色法的RSD值均小于10.5% ,因此更为稳定。在回收试验中,把已知量的致癌染料标准液根据实际染色情况添加到不同纤维的白色坯布上:直接蓝6、直接红28和直接黑38添加到棉织物上,酸性红26添加到毛织物上,碱性红9和碱性紫14添加到腈纶织物上,分散蓝1、分散橙11和分散黄3添加到涤纶织物上,并按照1.
3节进行前处理,提取的染料按1.4节测定。在5、50、500mg/kg的加标水平下,各种染料的回收率均在92.3%~106.8% ,且在提取过程中未发生染料的分解,
10次加标回收的RSD 值也均小于10.0%,表明本方法准确可靠。
参考文献:
[1] Test
Institute of the International Oeko-Tex
Association. General and special conditions[EB/OL]. http://www.Oeko-tex.com/OekoTex100_PUBLIC/,2008-01- 07.
[2] 王建平,苏红伟,洪晨跃,等.纺织品上致癌染料的检测方法研究[J]印染,2007(1):32-37
[3]范雪荣,纺织品染整工艺学[M]北京,中国纺织出版社,1996
[4]Zhang
X, Laursen R A. Development of mild extraction methods for the analysis of
natural dyes in textiles of historical interest using LC-Diode array detector2MS[J] Analytica chemistry,2005(77);2022-2025
[5]laing
David K, Richard Gill, Gandida Blacklaws. Characterisation of acid dyes in
forensic fibre analysis by high2performance
liquid chromatography using narrow-bore
columns and diode array detection [J]Journal of
chromatography,1988(442):187-208
[6]WhealsB
B, White P C, Paterson MD. High2performance
liquid chromatographic method utilizing single or multi2wavelength detection for the comparison of disperse dyes extracted from
polyester fibres [J]
Jouranl of
chromatography,1985(350):205-215
[7]Thorburn
BurnsD, McGuigan A A. The identification and determination of cationic dyes in
dye liquors for acrylic fibre and extracted from finished p roducts [J]
Fresenius Journal of Analytical Chemistry,1991(340): 377-379
[8]FuhM F,
Chia K J. Determination of sulphonated azo dyes in food by ion-pair liquid chromatography with photodiode array and
electrosp ray mass spectrometry detection[J]Talanta,2002(56):663-671