自乳化水性聚氨酯涂层剂自交联行为的研究
林萍 周宏(音) 中国纺织大学
原载:染整涂层及其他整理学术论文、资料选辑/1990
提要
本文采用聚醚二醇-异氰酸酯预聚体与多元胺伸链的方法,依靠聚乙二醇的亲水性制成自乳化的聚氨酯水分散液,并加入潜在自交联剂环氧氯丙烷以改善其各项性能。
本文运用经典方法对聚醚二醇与异氰酸酯的预聚反应进行了动力学研究,并通过近代物理及化学测试方法详细研讨了潜在自交联剂环氧氯丙烷和共聚醚中的环氧乙烷的含量对水性聚氨酯涂层的性能影响。
在合成聚氨酯涂层剂的基础上,还进行了涂层的应用工艺及其性能的测试。
一、引言
涂层织物是一种具有多功能,多用途的产品,在现代科学技术发展的推动下,涂层织物的研究开发方兴未艾,新产品层出不穷,其中最令人瞩目的要算聚氨酯涂层织物,它以其独特的性质引起了国内外研究人员的普遍关注。聚氨酯涂层剂有很多优点,如涂层薄、弹性好、粘着性好、手感柔软、
耐溶剂、耐低温、耐磨、防水透湿等,因此它的发展前景一定非常广阔。
水分散性聚氨酯的研究在国外早在1942年就己开始,七十年代初期就以商品的形式出现,到了七十年代后期,产品就更多了,如西德Bayer公司的Impranil系列等。而国内的研究还刚刚起步,用于织物涂层的聚氨酯水分散涂层剂目前在市场上尚属空白。为了开发这一具有很好发展前途的纺织新产品,各方面都很重视。国内已有好多单位在对水分散型聚氨酯进行研究,并取得了一定成果。我们应化研究室对聚醚型的水分散聚氨酯涂层剂进行了系统的研究,包括阳离子型、阴离子型和非离子型水分散聚氨酯。本文主要对非离子聚氨酯乳液进行详细深入的研究,探讨了由于潜在自交联剂的引入以及采用不同的共聚醚而引起的聚氨酯弹性体性能的变化规律。并运用动态粘弹谱以及一些经典分析的方法,对所合成的大分子结构特征进行分析,阐明了大分子的结构与其性能的关系。
二、实验材料与方法
(一)合成方法
自交链自乳化水性聚氨酯涂层剂的制备按Kazuo,Matsuda等[1]所研究的方法进行。
(二)制样方法
将上述乳液平铺在水平的玻璃板上,室温放置48小时自然晾干,然后在热风烘箱中按所需温度、时间焙烘,然后将薄膜缓缓剥离。
(三)测试方法
1、薄膜应力-应变曲线的测定;在DCS-500电子强力拉伸仪上进行,拉伸夹距为10毫米,拉伸速度为100毫米/分。
2、薄膜弹性回复率的测试;在lnstron 1122拉伸仪上进行,将原夹距为10毫米的试样以100毫米/分的速度拉伸至40毫米,除去外力,任其自然回复1O分后再拉伸。
3、薄膜应力松驰的测定;在Instron 1122拉伸仪上进行,夹距为10毫米,拉伸速度为100毫米/分,拉伸至400%伸长率,仃止拉伸,然后以20毫米/分的速度自动记录。
4、薄膜动态粘弹谱的测定;将薄膜放入DDV-Ⅱ-EA型动-态粘弹谱仪中,用液氮作制冷剂,频率为110Hz,升温速率为2℃/分,温度测量范围为-6O℃-l70℃。
5、涂层织物耐水压的测试;按常规在水压计上进行。
6、涂层织物透湿性的测试;按国家标准[2]进行,温度为40℃,相对湿度为90%。
三、实验结果与讨论
(一)预聚反应动力学
我们用2,4-甲苯二异氰酸酯(TDI)和二端羟基聚醚以2:1的摩尔比在催化剂作用下进行反应,其反应式为[3];
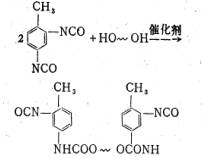
图1是预聚反应时异氰酸酯基的重量百分浓度随时间的变化曲线。当反应完全时,预聚体中异氰酸酯基的百分含量应为;
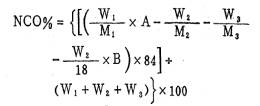
式中;W1――TDI重量(克)
W2――PG (共聚醚)重量(克)
W3――PEG(聚乙二醇)重量(克)
M1――TDI分子量
M2――PG平均分子量
M3――PEG平均分子量
A ――TDI的有效成份;
B――PG的含水量。
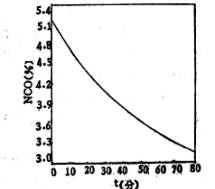
图l NCO%随时间的变化
根据反应配方,由上式可求得预聚反应完全时,理论NCO%=3.16%。
将图l的NCO%值取对数,作ln(NCO%)~t图,得到了两条不同斜率的直线,如图2所示,由于共聚醚的端羟基有伯羟和仲羟两类[4],伯羟基比仲羟基空间位阻小,因此反应活性大。由图2的直线可以认为,端羟基聚醚与TDI的反应为假一级反应,伯羟基与TDI的反应速度大于仲羟基。M.Kaplan等人[5]曾测得芳环异氰酸酯与醇的反应为一级反应;R.G.Ferrillo[6]用恒温DSC方法研究端羟基聚醚与TDI的反
应,也得到一级反应的结果。那么,可得
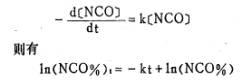
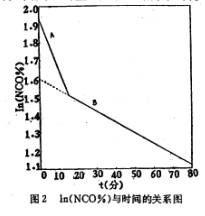
图2 ln(NCO%)与时间的关系图
这样,由图2的直线A、B的斜率可得在该反应温度下伯羟基与TDI的反应速率常数k1=kA-kB=2.08×1O-2(分-1);仲羟基与TDI的反应速率常数K2=KB=6.05×10-3 (分-1)。k1/k2=3.43,即伯羟基与TDI的反应速率约为仲羟基的三倍,这与文献报导的结果相符[3]。
(二)聚氨酯大分子结构与其动态粘弹性能的关系
1、大分子结构对其玻璃化转变温度的影响
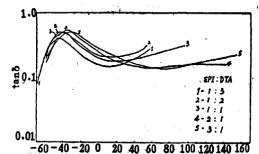
图3表示聚氨酯薄膜的衰减随引入的交联基团含量的变化情况,从图中曲线可明显看出,随着交联基团含量的增加,Tg向高温侧偏移,衰减峰的宽度亦变宽,这与文献报道是相符的[7-9]。这主要是由于潜在自交联剂环氧氯丙烷的引入,增加了分子间的氢键及共价交联,限制了链段的热运动,使
Tg升高。所以随着环氧氯丙烷含量的增加,聚氨酯薄膜的Tg上升。具体数据见表1
T(℃)
图3不同EPI/DTA克分子比的薄膜的衰减一温度关系图
表1 交联基团含量对薄膜Tg的影响
|
EPI:DTA |
1:3 |
1:2 |
1:1 |
2:1 |
3:1 |
|
Tg(℃) |
-45.3 |
-42.0 |
-41.2 |
-35.4 |
-32.4 |
注:EPI-环氧氯丙烷; DTA-二乙烯三胺
图4是共聚醚中不同环氧乙烷(E0)含量对聚氨酯薄膜衰减的影响。当EO%提高时,大分子链上-CH2CH2O-基的含量增
多,醚键的增多使得大分子链的柔性大大增加,链段的运动阻碍变小,Tg下降[10]。表2是不同环氧乙烷含量的聚氨酯薄膜的Tg数据。
|
|
|
图4 不同EO含量的薄膜的衰减-温度关系图 |
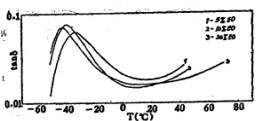
表2 共聚醚中EO%对薄膜Tg的影响
|
E0% |
5% |
10% |
20% |
|
Tg(℃) |
-33.0 |
-38.8 |
-42.0 |
2、大分子结构对其动态模量的影响
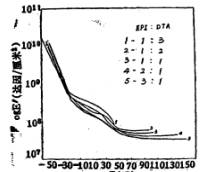
图5是不同交联基团含量的聚氨酯薄膜的动态模量-温度曲线。从图中曲线可看出,在温度较低(低于Tg)时,交联基团的含量对其动态模量的影响甚微;当温度达玻璃化转变时,在很小的温度范围内模量下降很快;当温度进一步提高时,随着交联基团含量的不同,其动态模量随温度的变化出现了
差异,对于交联基团含量很少的聚氨酯大分子,由于出现了分子键的流动。模量随温度进一步下降(图5曲线1),而对于交联基团含量较高的聚氨酯大分子,当温度达到一定程度时,其模量不再依温度的变化而变化,这时,薄膜处于橡胶态,而且随EPI含量的增加,其橡胶态的持续温度范围越广 (图5曲线2→5)。这是环氧氯丙烷含量的增加,使分子间的共价交联和氢键增加的缘故[11]。
|
|
|
T(℃) |
|
图5 不同EPI/DTA克分子比的薄膜的动态模量-温度曲线 |
(三)聚氨酯大分子结构与其力学性能的关系
1、大分子结构对薄膜拉伸性能的影响
图6是不同交联基团含量对聚氨酯薄膜拉伸性能的影响。由图可见,薄膜的断裂应变(εb)随着交联基团含量的增加而下降,初始模量(Er)随着交联基团含量的增加而增加,断裂应力(σb)则在EPI/DTA为l/2时达到最大值。
环氧氯丙烷的引入增强了分子间的氢键作用,并在分子间产生共价交联,使链段的运动受到限制,所以随着环氧氯丙烷含量的增加,εb相应降低,Er则增大。而σb存在一个最大值是由于EPI含量过高时,分子链间的共价交联过度,阻碍了链段的取向,容易产生应力集中,此外,还使分子链的侧基增多,势必影响分子链的紧密排列,使分子间的氢键作用不能充分发挥,因而σb反而下降。由图6所得具体数据见表3。
|
|
|
图6不同EPI/DTA克分子比对薄膜拉伸性能的影响 |
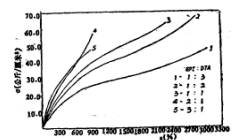
表3不同EPI/DTA克分子比对薄膜拉伸性能的影响
|
EPI:DTA |
l:3 |
1:2 |
1:1 |
2:1 |
3:1 |
|
σb(公斤/厘米2) |
47.0 |
66.3 |
63.9 |
57.7 |
47.8 |
|
εb(%) |
3060 |
2856 |
2318 |
933 |
908 |
|
Er(公斤/厘米2) |
9.4 |
10.0 |
10.8 |
13.6 |
13.2 |
图7表示共聚醚中不同环氧乙烷含量对薄膜拉伸性能的影响,由图可见,环氧乙烷含量的增加使薄膜的断裂应力(σb )和初始模量(Er)增加,而使断裂应变(εb)减小。
共聚醚中环氧乙烷含量的增加,会使大分子的微相分离程度减小,这样,起物理交联作用的硬段区域分布在软段相中的数目就增多。在外力的作用下,不容易发生形变。
因此,随着环氧乙烷含量的增加,断裂应力和初始膜量增加,而断裂应变则下降。由图7可得数据列于表4。
|
|
|
t(%) |
|
图7 不同EO含量对薄膜拉伸性能的影响 |
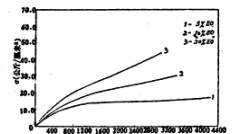
表4 不同EO含量对薄膜拉伸性能的影响
|
E0% |
5% |
10% |
20% |
|
σb(公斤/厘米2) |
17.2 |
30.3 |
44.1 |
|
εb(%) |
4211 |
3412 |
3035 |
|
Er(公斤/厘米2) |
7.0 |
7.9 |
8.8 |
2、大分子结构对薄膜弹性回复性能的影响
聚氨酯弹性回复率的大小,主要取决于永久形变的大小,永久形变越大,弹性回复率就越小。
表5 不同EPl/DTA克分子比对薄薄弹性回复率的影响
|
EPI:DTA |
l:3 |
1:2 |
1:1 |
2:1 |
3:1 |
|
弹性回复率(%) |
71.0 |
74.7 |
78.0 |
84.3 |
86.3 |
表5是不同交联基团含量对薄膜弹性回复率的影响。显而易见,随着交联基团含量的增加,弹性回复率增大。这是由于环氧氯丙烷含量增加时,分子间的共价交联增多,这就便链段的伸展和整个分子间的相对滑移受到限制,因此永久形变减小,弹性回复率增大。
表6 不同EO含量对薄膜弹性回复率的影响
|
E0% |
5% |
10% |
20% |
|
弹性回复率(%) |
90.0 |
87.7 |
85.3 |
表6是共聚醚中不同环氧乙烷含量对薄膜弹性回复率的影响,当环氧乙烷含量增加时,薄膜的弹性回复率降低。这是因为当环氧乙烷含量较少时,软、硬段之间的微相分离较大,类似于网状结构中的网眼较大,软段相就容易发生形变,当薄膜受外力作用时,硬段被拉伸而产生相对滑移的可能性就比较小,从而使硬段中氢键被破坏而产生永久形变的可能性亦较小,因此弹性回复率较高。
3、大分子结构对薄膜应力松弛性能的影响
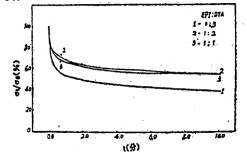
图8是不同交联基团含量的薄膜应力松驰率(σt/σ0)随时间变化的规律。应力松弛率就是在薄膜被拉伸到400%后恒定应变,不同时刻的应力(σt)与原应力(σ0)的比值(σt/σ0)
。σt/σ0值越大,应力松弛越慢。从图中曲线可看出,当环氧氯丙烷的含量从EPI/DTA=1/3增加到1/2时,应力松弛大大减慢。这是因为环氧氯丙烷含量的增加,使得分子间的氢键密度和共价交联增加,这就限制了链段的运动和整个大分子链的相互滑移,回复到平衡态的速率就较慢,故而应力松弛减慢。当环氧氯丙烷的含量从EPI/DTA=l/2增加到1时,应力松弛反而有所加决(图中曲线2→3)。这是由于环氧氯丙烷含量过高,共价交联的增多使应力容易集中;侧基的增多也使大分子结构变得疏松,分子链容易顺着外力方向运动回复到平衡态,以消除内应力,所以抵消了由于共价交联增多而使应力松弛减慢的效应,使得应力松弛反而有所加快,但加快不多。
图9是共聚醚中不同环氧乙烷含量对薄膜应力松弛的影响。从图中曲线可看出,当环氧乙烷含量增加时,薄膜的应力松弛减慢。
|
|
|
|
图8 十分钟内不同EPI/DTA克分子比对应力松弛的影响 |
图9 十分钟内不同EO含量对应力松驰的影响 |
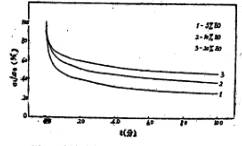
随着共聚醚中环氧乙烷含量的增加,聚氨酯大分子中-CH2CH2O-基的含量增多,这样,硬段中的-NH2基与-CH2CH2O-基中的0原子形成的氢键增多,这就限制了链段的运动和大分子链的滑移。而应力松弛主要就是在薄膜拉伸到400%时恒定应变,借助于链段的热运动使之回复到平衡态以减少或消除内应力。这样,环氧乙烷含量的增高,使链段顺外力方向运动以减少或消除内应力变得困难,应力松弛减慢。
(四)聚氨酯大分子结构与其涂层织物性能的关系
1、大分子结构对涂层布耐水压性能的影响
表7 不同EPl/DTA克分子比对涂层布耐水压性能的影响
|
EPI:DTA |
0 |
1:3 |
1:2 |
1:1 |
|
耐水压(毫米水柱) |
370 |
430 |
450 |
500 |
从上表数据可看出,随着交联基团含量的增加,涂层布的耐水压值升高。这是因为交联基团含量的增加,使涂层布上的聚氨酯膜在焙烘过程中产生的共价交联亦增多,导致交联密度增加,使涂层布的耐水压值提高。
2、大分子结构对涂层布透湿性能的影响
表8 不同EPl/DTA克分子比对涂层布透湿性能的影响
|
EPI:DTA |
O |
1:3 |
1:2 |
1:1 |
|
透湿量(克/米2・24小时) |
3156 |
2932 |
2562 |
2512 |
上表数据表明,交联基团含量增高,涂层布的透湿量下降。这是因为环氧氯丙烷的引入使涂层膜在焙烘过程中产生化学交联,封闭了部分亲水基团,使涂层膜的亲水性下降,导致透湿量降低。此外交联作用还限制了链段的热运动,这也会导致透湿量降低。
四、结论
通过以上讨论,得出以下结论;
1、2,4-甲苯二异氰酸酯与聚醚二醇的反应为假一级反应,聚醚二醇中伯控羟基和仲羟基与TDI的反应速率常数分别为;
2.08×10-2分-1和6.05×10-3分-1。
2、环氧氯丙烷的引入对聚氨酯薄膜的性能产生较大的影响,其含量的增加引起薄膜的Tg升高,橡胶态明显,断裂伸长降低,初始模量升高,弹性回复率增加;而薄膜的断裂应力和应力松弛均存在极值,交联基团含量过高,会使聚氨酯大分子发脆,所以环氧氯丙烷的含量最佳应为EPI/DTA克分子之比为1/2。
3、环氧氯丙烷的引入还影响涂层织物的性能,其含量的增加使涂层织物的耐水压值升高,透湿量降低。
4、共聚醚中环氧乙烷含量的增加会引起聚氨酯薄膜的Tg下降;并使薄膜的断裂应力和初始模量增高,断裂伸长下降,弹性回复率下降,应力松驰变慢。
参考文献
[1]
US Pat. 4012349
[2]
GB 1037-30
[3]
H.F.Mark, N.G.Gayland and N.M.Bikalcs,《
Encyclopedia of polymer Science and Technology》,
11(1969), 506.
[4]
H.F.Mark, N.G.Gayland and N.M.Bikales,《
Encyclopedia of polymer Science and Technology》,
6(1976), 145.
[5]
M.Kaplan,《J.Chem.Eng.Data》,6(1961),272.
[6]
k.G.Fcrrillo,V.D.Arendt and A.H.Granzoro,《J.Appl.Polym,Sci.》,28(1983),2281-2289.
[7]
T.Murayama and J.P.Bell,《J.Polymer Sci.》,8(1970), 437, A2.
[8]
D.Katz and G.Salee,《J.Polymer Sci.》, 6(1968),801,A2.
[9]
G.Pohl and S.Kastner,《J.Polymer Sci.》, C16(1968) ,4133.
[10]
CHIN-PING YANG and WEN-LIN WU,《J.Appl.Polym.Sci.》,28(1983), 2509-2525.
[11]兰立文,《高分子物理》,1985