ʯīϩ����:����������ʯīϩ��ֱ������yd20824
����#1��������#1���ᄌ��1������1,2�����ҷ�*1 1��������ѧ��ѧ����ӹ���ѧԺ������ѧ����ѧ�о����ģ�����100871��2��������ѧ��ѧԺ������100871��
�ո�: 2015-10-15,�� ��: 2015-11- 12,�� �������: 2015-11- 13,
�����ص�����о���չ�滮��Ŀ(973) (2013CB932603, 2012CB933404, 2011CB921903, 2013CB934600),������Ȼ��ѧ����(51432002,51290272, 51121091, 51222201, 11222434), ������(20120001130010)�ͱ����п�ѧ����ίԱ��(Z151100003315013)����
#ͬΪ��Ŀ������
*ͨѶ����. Email: zfliu@pku.edu.cn�� Tel: +86-10-62757157.
ԭ�أ���������ѧѧ���� 2016/1��14-27
��ժҪ��������һ����ʷ�ƾá���;�㷺�����ι����β���,��ʯīϩ���ǽ��������ֵĽ���̼ԭ����ɵĶ�ά��״���ϡ�ʯīϩ���г��ߵĻ�еǿ�ȡ������ԡ������Ժ�����,ǡ���봫ͳ�IJ����γɻ�������ʯīϩ�벣�������һ��,�ڱ������ԵĻ�����,ͬʱ������ͨ���������ԡ������Ժͱ�����ˮ��,���зdz���Ҫ��ʵ����������ۼ�ֵ�������Һ��ͿĤ����ת�Ƶķ���,ֱ���ڲ�����������ʯīϩ�ܹ��Ӹ����ϱ���������Ⱦ�����������ʯīϩ���ܵ��½�,�Ӷ���չ��һ�����Ͳ�������ʯīϩ���������Ľ����������о����ڸ��ֲ�������ֱ������ʯīϩ���о���չ,���а���ʯīϩ�ڹ�̬���²���������̬��������ĸ�������,�Լ����õ������帨���ֶ�ʵ��ʯīϩ����ͨ��������ĵ�������,���Դ�Ϊ������չ�����ֻ���ʯīϩ������Ӧ��ʵ�����ܽ�չ����ʯīϩ�������Ʊ���Ӧ�õ�δ����ս�뷢չ����
���ؼ��֡�ʯīϩ����̬����������̬��������ѧ����������������帨��
����ͼ����š�TQ171.1 O647 doi��10.3866/PKU.WHXB201511133
1
����
�������ķ�չ�Ϳ�ѧ�����Ľ������벻�����ϵķ�չ��ÿһ����Ҫ�����߽����ǵ�������Ƕ����ʽ���ش����������һ�����͵����ӡ���Ϊһ�ֵ��͵Ĵ�ͳ���ϣ�������ʹ�ÿ����ݵ���Ԫǰ��ֱ�����죬�����Ѿ���Ϊ���������в��ɻ�ȱ����Ҫ���ϣ����������õ����Ⱥ͵����ijɱ������㷺Ӧ���ڽ��������������ӡ���ѧ��ҽҩ�Լ�ʳƷ���������Ȼ�����������ĵ����Ժ͵����Զ��ܲ��Լ�����ڸ��������Ӧ�á�����һ��ͬ�����и����Ե����Ͷ�ά���ײ��ϡ���ʯīϩ����2004����Ӣ������˹�ش�ѧ��Novoselov���״����ý������뷨�Ʊ�����֮��[1]��������о�����ʮ���ȡ����ͻ���ͽ��ķ�չ[2-9]��������Ϊ21������ߴ����Ե��²��ϡ�����ʯīϩ�������������������ʣ��糬�ߵĻ�еǿ��[10]�����ߵ�������Ǩ����(��߿ɴ�200000 cm2��V�C1��s�C1)[11]���ȵ���[12]������ĵ�����1������7�ȣ�ʹ�����������籡Ĥ[13]������������[14]����ЧӦ�����[15]�����̽��[16-18]������ӵ��[19]��DNA����[20-21]�����������ﴫ����[22-23]�����ܸ��ϲ���[24]������������Ź�����Ӧ��ǰ����
ʯīϩ�����������������Ϊ����Ӧ������ķ�չ�춨�˻�����Ȼ���䳬���Ķ�ά�ṹ������ʯīϩ���ѱ�����ʹ�ã���Ҫ������ij�ֳĵײ��ܱ��ֳ���Щ���ܡ��������к�ʯīϩ��ƥ������ԣ�ͬʱ�ڵ����ԡ������Ժͽ����Է����ܹ���ʯīϩ�γɻ�������ˣ���ʯīϩ�벣�������һ�𣬷�չ��һ�������ϲ��ϣ���ʯīϩ���������ܹ����ֲ����������Ժõ��ŵ㣬���ܹ���ʯīϩ�����еij��ߵ����ԡ������Ժͱ�����ˮ�Ե��ص㸳�貣����ͬʱ���ڲ����ɱ�������Ӧ�ù㷺��ʹ��ʯīϩ����������Ϊһ���ܹ��߽��ճ���������Ͳ��ϡ�
��Ҫʵ��ʯīϩ�����IJ�ҵ��Ӧ�ã�������Ҫ����ı��Ǹ�Ʒ��ʯīϩ�����Ĵ��ģ�Ʊ����⡣����Ϊֹ���Ʊ�ʯīϩ�����ķ�����Ҫ�����֣���һ������Һ�����ʯīϩ��ԭ����ʯīϩ��Ϊԭ�ϣ�ͨ��Һ��ͿĤ�ķ����ڲ�������Ϳ��һ��ʯīϩ��Ĥ[25-26]��������Ի�ѧ�������(CVD)���ڽ�������(��ͭ)������������ʯīϩΪԭ�ϣ�ͨ��ת�Ƶķ�����ʯīϩת�Ƶ���������[27-28]��Һ��ͿĤ����һ�ֽ�����õ�ʯīϩ�����Ʊ�������ͨ��Langmuir-Blodgett (LB)Ĥ������Ϳ����Ϳ���ֶν�ʯīϩ���ǵ��������γ�ʯīϩ��Ĥ��Ȼ������ʯīϩƬ�ijߴ�С��ȱ�ݶ࣬���������������������ַ����Ʊ���ʯīϩ��Ĥ�ľ����Ժ�Ʒ�ʶ��ܲ���������ܴ��ںܴ��ࡣ��CVD���ڽ�������������ʯīϩ��������ת�ƣ�����һ�ֱȽ�ͨ�õķ���������CVD�������м۸�������Ʊ����ɿ���ǿ�����ƣ�ͬʱ���ڽ�������ʯīϩ�������������ԵĴ�����[28-31]���������CVD�����ڽ�������������ʯīϩ�ѳ�Ϊʵ�ִ��������Ʒ��ʯīϩ�Ʊ�����������[32-36]�������о���Ҳ�ڽ�������ʯīϩ�Ŀɿ��Ʊ����������˴���Խ�Ĺ���[3,27,37-40]��Ȼ�����ڽ��������ױ���������ʯīϩת�Ƶ���������Ĺ����У�����Һ�ѧ�Լ���ʹ�ã���ɱ���ش���ʯīϩ����Ⱦ���⣬�Լ�������ת�ƹ��շ�������������ʯīϩ��ȱ�ݺ�����ȣ���Щ���ض��������Լ������õ�ʯīϩ�ĸ������ܡ�
���������Ʊ�ʯīϩ�����ķ������õĶ��Ǽ���Ʊ��������ȵõ�ʯīϩ�ٽ�ʯīϩ�����ڲ������棬�������������ɱ��߰��������⣬�õ���ʯīϩ��Ĥ���������ܵ������ص�Ӱ�졣 ��ˣ�����ܹ�ֱ���ڲ�������ɿ��Ʊ������������������ʯīϩ��Ĥ���Ϳ��Էdz���Ч�ر���ͿĤ��ת�ƹ��̴�����������⣬�����ܹ�������������ɱ��������ڹ�ҵ�������������������ȣ�����CVD�����ڲ�������ֱ������ʯīϩ��Ҫ����������������ѣ�һ���沣�������Ĵ����ܺ�����ʹ��̼Դ�ѽ����ѣ�ʯīϩ�ڲ�������ijɺ˺��������ѽ��У���һ���沣���������¶�һ�㶼����ʯīϩ�������¶ȣ�ʹ�ò�������ʯīϩ��������Ҫ�����辶����ǰ�������о����Ѿ���ʼ���ڲ�������ֱ������ʯīϩ���о���Ϊʯīϩ�������Ʊ���չ��ϵ�з���[41-43]�����Ľ�����Щ�о��ɹ������ܽᣬϵͳ���������ڲ�����������ʯīϩ������������̽����
|
|
|
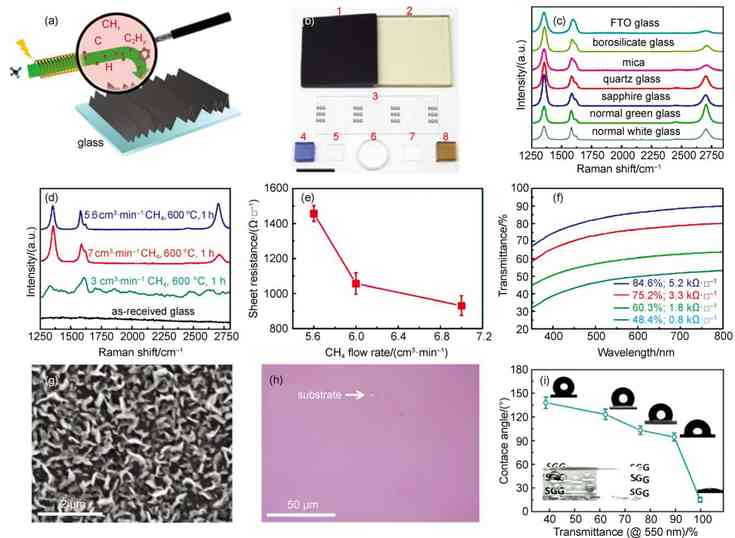
ͼ1 ʯīϩ�����²�������ĸ���ֱ������[41] |
|
(a) schematic of graphene growth on solid glass by catalyst-free atmospheric-pressure chemical vapor deposition (APCVD) route�� (b) scanning electron microscope (SEM) image of directly grown APCVD graphene on quartz glass�� (c) optical microscope (OM) image of the transferred graphene film onto the SiO2/Si substrate�� (d) photograph of the borosilicate glass substrates before (leftmost) and after graphene growth with different CH4flow rates at 2, 5, 7.5, and 10 cm3��min�C1�� (e) representative Raman spectra of directly-grown graphene on different solid glasses. |
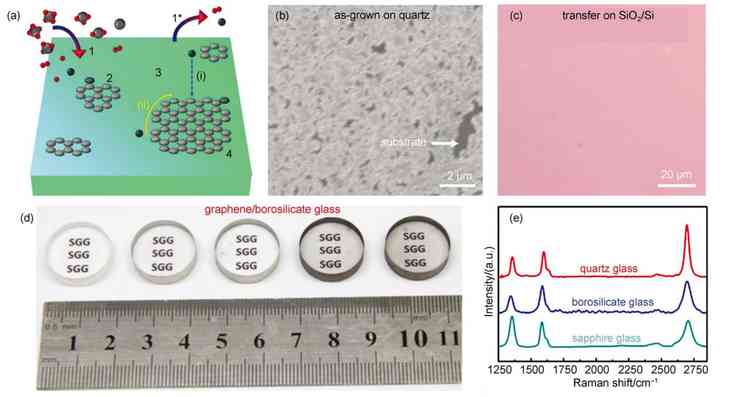
2
ʯīϩ�����²������������
������CVDֱ�������ķ������Լ���Ϊ̼Դ����������Ʒ�ʵ�ʯīϩͨ����Ҫ1000�����ϵĸ��¡�Ϊ���ڲ�����������ʯīϩ������Ҫѡ�����²�����Ϊ�������ס�����ͨ�ƸƲ�����ȣ����²����������¶ȸ���1000����Ϊʯīϩ�ڹ�̬��������ĸ��������ṩ�˿����ԡ���ˣ���������ѡ����ʯӢ��������貣��������ʯ�����²�����Ϊ���ף�ֱ����������������ʯīϩ��
������ʯīϩ�������зdz���Ҫ�����ã���Ҫ�������������棺���״�̼Դ�ѽ��������������̼Ǩ�Ƶ������Լ�ʯīϩ�ڻ����ϳɺ˵������������������ȣ����������Ծ��з����Ժͱ����ԵĹ��ۼ�Ϊ�������̼�����������ڱ����γ����õĻ�ѧ��������������������̼Դ�ѽ�Ĵ����÷dz����ޡ���һ���棬����̼ԭ���ڲ��������Ǩ������ҪԶ���ڽ������棬�⼫���������̼�����ڲ��������Ǩ�ơ���ˣ��������ȣ��ڲ��������ʯīϩ�����ձ���ֳ��ϵ͵��������ʺͽϲ�Ľᾧ������
ʯīϩ�ڹ�̬�������������������ͼ1(a)��ʾ�����ڲ�������̼�⻯����ֽ�Ĵ�����ʮ�����ޣ����ѽ��Ϊ�����ѽ����Ҫ��ʽ����Ҫע����ǣ��ڵ�ѹCVD (LPCVD)��ϵ��̼Դ������ӵ�Ũ�Ƚϵͣ�����Ǩ��Ѹ��[44-46]��������ʯīϩ�ڲ�������ɺ˺�������Ҫ�����ͨ�����õĶ��dz�ѹCVD (APCVD)��ϵ[47-53]������������̼����Ũ�ȴﵽ������״̬ʱ������ڻ���λ�㴦�ɺˡ����ڲ��������Ǩ�����ݺܸߣ�ʯīϩ��Ǩ�������ܵ����ƣ��ѽ��̼����ֱ���䵽ʯīϩ���ı�Ե����Ϊ��������ʯīϩ������һ����Ҫ��ʽ����Ҳ������������ʯīϩȱ�ݽ϶ࡣ����ʱ������ƣ���Щ������ʯīϩ����ƴ����һ���γ�������ʯīϩ��Ĥ����ͼ1(b��c)��ʾ��������������һ����Ҫ2�C6 h��ʯīϩ�����ߴ�һ���ڼ����������ҡ���ѹCVD��ϵ�²�������ʯīϩ�������ܵ�̼ԴŨ�ȵ�Ӱ�죬��ͼ1(d)��ʾ����������Ũ���ܹ���Ч����ʯīϩ�IJ��������볣ѹ�½�����������ʯīϩ�Ľ��������[44,54]�����ü�����Ϊ̼Դ�����dzɹ�����ʯӢ��������貣���Լ�����ʯ�����ı�����������Ʒ�ʽϸߡ����ȶȺõĵ���ʯīϩ��Ʒ����ͼ1(e)��ʾ����������Ǩ���ʿɴ�553�C710 cm2��V�C1��s�C1��
�¶ȶ�ʯīϩ�ڲ������������кܴ�Ӱ�죬��ͼ2(a��b)��ʾ�����������¶��ܹ��dz���Ч������ʯīϩ��Ʒ�ʡ�һ����������Ҫ�ѽ�;�������ѽ⣬�¶�Խ�����ѽ��Խ���ף�����̼���ֵ�Ũ��ҲԽ�ߣ���һ���棬�¶�Խ����̼����Խ���˷���������Ǩ�����ݣ�����Ǩ�����ʣ�ʯīϩ�ij���Ҳ��Խ����ˣ��ڸ����¿�����Ч���ٲ�������ʯīϩ��ȱ�ݣ����ʯīϩ��Ĥ�������;����ԡ����¶��⣬����ʱ��Ҳ��Ӱ�첣��������ʯīϩ��������Ҫ���ء���ͼ2(c��d)��ʾ���ӳ�����ʱ��һ�����ܹ�����ʯīϩ��ȣ���һ�����ܹ�����ʯīϩȱ�ݡ����ڲ��õ��dz�ѹCVD��ϵ������ʱ�������̼��Ƭ���������ڲ������棬����ʯīϩԽ��Խ���������������ЧӦ[44]��ͬʱ�������˻��ܹ�ʹ̼ԭ�ӵ��Ų�����������������ʱ����ӳ�ʯīϩȱ�ݻ�õ�һ���̶ȵ�����
|
|
|
ͼ2 ʯīϩ�����²����������������[41] |
|
(a)Raman spectroscopy of the temperature-dependent growth of graphene on sapphire glass�� (b) corresponding band (D, G, 2D) intensity ratios (ID/IGand I2D/IG) as a function of growth temperature extracted from (a)�� (c) Raman spectroscopy of the time-depending growth of graphene on quartz glass��(d) Corresponding ID/IGand I2D/IGratios as a function of growth temperature extracted from (c). All the Raman spectra are normalized to G peak intensity. |
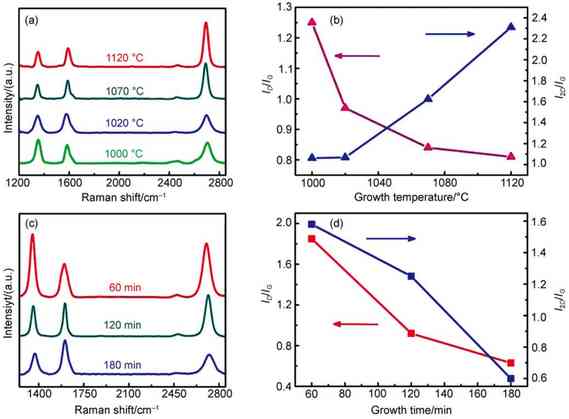
ʯīϩ�����²�������������������ø���ֱ���ѽ�̼Դ֮�⣬�о���ԱҲ�������ý���Զ�̴������ڲ���������ʯīϩ��Chiu�о���[52]����ͭ��Զ�̴�������ʯӢ����������ʯīϩ��������ʯӢ�������εĺ���λ�÷���һ��ͭƬ��������ͭ�����ڸ����´�������ѽ⣬�õ���̼��Ƭ�����ε�ʯӢ��������ɺˡ�������Choi�о���[53]�ڴ˻����Ͻ����˸Ľ�����ͭƬ����ʯӢ�������Ϸ��������Ӵ�����ʯӢ��������������ȱ�ݷ��С�ĵ���ʯīϩ��Fu�о���[55]�����ش���ͭ����Զ�̴�����Ϊ��ͬ�������ص�����ѹǿ��ͭ��ʮ���������ζ�Ÿ�����ز��뵽����Ӧ�У������������̼Դ���ѽ��ٶȡ������������ַ�����ʯӢ�����������������ȵĵ���ʯīϩ�������ij�������Ч�����ԡ���Ȼ������Զ�̴������ܹ����̼Դ���ѽ�Ч�ʣ��ӿ����������̣������ַ���ʼ������ȫ��������IJ������⡣���⣬���ڽ��������ڿռ�ֲ������ȣ�����������ʯīϩ�ڴ�߶��Ϻ�Ȳ�����
3
ʯīϩ������̬�������������
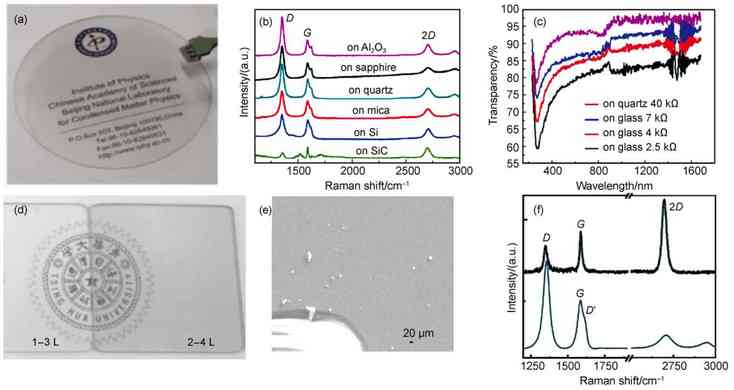
��ͨ����(һ��ָ�ƸƸ�������)�������¶�һ����600�����ң�����1000�����������������ʯīϩ�ڴ�ͳ�����в������С�Ȼ�������������ڽ�������������Ʒ��ʯīϩ������½����չ[56-59],Ϊ���ǽ�ʾ��һ����ͨ���������������ʯīϩ�Ŀ����Ե�·������������̬��������ֱ������ʯīϩ����Ϊ��Ҫ���ǣ����̬������ȣ�����̬�����ı���߶Ⱦ�һ���Լ�Һ����еĴٽ�̼Ǩ�Ƶ�����[58,59]��ʹ��ʯīϩ������̬��������ijɺ˺��������̴��ڽϴ�����ơ�
����̬���������ʯīϩ������Ȼ���ó�ѹCVD��ϵ����1000���������˻�30 min�Ի���ȶ�������̬���������ͨ�������Ϊ̼Դ��������ѽ���Ȼ�����ѽ�Ϊ��������ʱ��һ�������2 h��ʯīϩ������̬�������������������ͼ3(a�Cd)��ʾ������һ�������������Ϊ�������ɺˡ����������ͻ������������Ρ������dzɺ˹��̣����������е�30 minʱ�γɿɼ���ʯīϩ���ģ�ʯīϩ�����ڲ���������ͬʱ�ɺ˵ģ��ں������������л������������µĺ��IJ�����Ȼ�����������̣���������һ����ʱ�����γɳߴ�ԼΪ1 ��m���ҵ�ʯīϩԲƬ����ЩԲƬ�Ĵ�С�ͷֲ���ʮ�־��ȣ�������Ϊ���ڲ��������Ǹ���ͬ�Եģ�ͬʱʯīϩƬ�����ڲ����������һ���̶ȵ�Ǩ����Ϊ����ͼ3(e�Ch)��ʾ��ͨ���߷ֱ������������(HR-TEM)��ѡ����������(SAED)�Լ��������ȱ����ֶ�֤ʵ��ЩԲƬΪ����ʯīϩ(���м�ɺ�����)�����Ҿ��нϸߵĽᾧ�Ⱥ�Ʒ�ʡ�ͨ���ӳ�����ʱ�����Ƿ��֣���ͬ�ڹ�̬�������ף�ʯīϩ�����ڲ���������������������������л���������������ֹͣ�������dz��������γɶ��ʯīϩ��ͼ3(i)�Ա�����ͬ������ʯīϩ�ֱ���SiO2/Si�ĵ�����̬���������������������ڲ�������90%���ϵ�ʯīϩƬֱ��λ��0.8��1.0 ��m֮�䣬�ҳߴ��С���У���SiO2/Si�����ʯīϩƬ�ߴ���ֱ���0.1��0.4 ��m֮�䣬��˲������������ķ���������һ���̶��ϻ������ڲ�������̼Ǩ������������ijߴ�С�����⡣
�����ἰ��ʯīϩ������̬��������������ٶȻ��Ż�����������ֹͣ����˵����ӳ�����ʱ��������Ч��ʹ��ƴ�ӳ�������Ĥ���������ȵġ�ͨ������£�����ͨ�����Ƽ���Ũ�Ⱥ������¶�����ò�ͬ���Ƕȡ���ͬ������ʯīϩ��Ĥ����ͼ4(a�Cd)��ʾ������ͨ�����Ƽ���Ũ�Ⱥ������¶��Լ�����ʱ�䣬ʵ���˶�ʯīϩ���dz̶ȵĿ��ơ�������Ũ�Ƚϵ�ʱ��ʯīϩԲƬ����ɢ�����������ż���Ũ�ȵ�������ЩԲƬ��ʼ�ۼ���һ�𣬽���ƴ�ӳ������ʯīϩ����ͼ4(e)��ʾ��������Ũ���㹻��ʱ�����ܹ��γ�˫������������ʯīϩĤ����ͼ4(f��g)��ʾ��
�¶ȶ���ʯīϩ������̬��������������Ӱ���Ƕ��ġ�һ���棬�¶�����������̼Դ���ѽ⣬ͬʱ����̼���������ڲ��������Ǩ��������������ʯīϩƬ���ø��������һ���棬ʯīϩ�ٽ�ɺ˳ߴ����¶����߶������¸����³ɺ���Ŀ���٣�ʯīϩԲƬ����ϡ�裻����֮�⣬�¶�Խ�ߣ�����̬���������˶�Ҳ��Խ���ң�����Ư����ʯīϩԲƬҲ��Խ��������ײ��Ҳ��Խ������������ǰ���ں���һ���γ�ʯīϩƬ�ľۼ��壬��ͼ4(h��i)��ʾ����Ҳ������̬�������������еġ�
�������¶Ⱥͼ���Ũ���⣬����Ũ��Ҳ���ʯīϩ������̬���������������������Ӱ�졣��ͼ4(j��k)��ʾ����������ռ����С��1% (2 cm3��min�C1)ʱ��ʯīϩ��������������������ռ��������25% (50 cm3��min�C1)ʱ��ʯīϩͬ�������������Ҳ���������ò��ʼ�����ı䡣������Ũ���ڴ�֮��ʱ������̬���������ܹ���������������ʯīϩ����ʱ��������ʯīϩ����������һ���Ĵٽ����ã������������������ܶ��������е��ڡ�
��ʯīϩ�������֮������̬������Ҫ���¹̻����õ���̬��ʯīϩ���������¹��̶��ڲ���������Ʒ��Ӱ��ܴ�Ϊ�����Ƿ��չ�ҵ�ϸ��������Ľ��¹��õ���ࡢƽ�����������ݺ;���������ʯīϩ������
|
|
|
ͼ3 ʯīϩ������̬��������ĸ���ֱ������[43] |
|
(a) schematic of graphene growth on molten glass�� (b�Cd) graphene disk sample grown at 970 ��C with the gas flow recipe Ar/H2/CH4: 150/20/10 cm3�� min�C1for (b) 30, (c) 120, and (d) 360 min, respectively�� (e) low-magnification TEM image of the graphene film, and the inset is SAED pattern�� (f)HR-TEM image of the universally existed monolayer�� (g) HRTEM image revealing good quality of produced graphene. Inset shows the atomically resolved STM image (Vs= 35 mV, It= 8 nA) of graphene honeycomb lattice. scale bar: 0.5 nm�� (h) Raman spectra of samples after experiencing control experiment (1020 ��C�� Ar/H2: 150/15 cm3�� min�C1�� growth time of 2 h) with/without the presence of CH4carbon feedstock (8 cm3�� min�C1)�� (i)comparison of the lateral sizes of graphene directly grown on molten glass (red column) and solid SiO2(blue column) substrates. Inset shows the corresponding SEM images of as-grown graphene on molten glass (top) and solid SiO2(bottom) substrates under identical conditions. scale bars: 1 ��m |
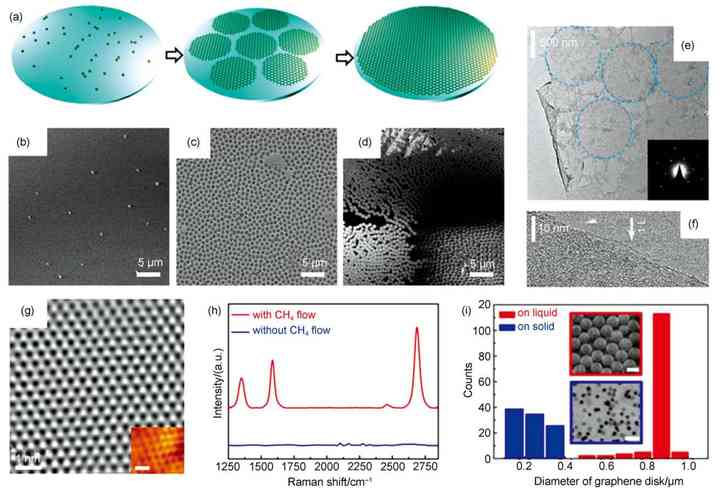
����������̬��������ʵ����ʯīϩ�Ŀɿ������������ʯīϩ�����Ĺ�ģ������������Ҫ���壬��Ϊ�ڲ�������������ͬ��������������̣���CVD������ʯīϩ�����������У�����ʵ��ʯīϩ�����������������⽫����ؼ��������̣���Լ�ɱ���
4
����������ʯīϩ�ڲ������������
��Ȼʯīϩ�ڸ��������µ�������������Ʒ�ʵ����������Ƕ�����Щ�Ѿ����͵IJ������������������ᵼ������ۺ����ʷ���������ת�ı仯�����ʵ�ֵ���������ʯīϩ�ڹ�̬��������Ŀɿ������Ƿ�չʯīϩ��������Ҫ��ɲ��֡��������帨����ѧ�������(PECVD)�����ǵ�������ʯīϩ����Ч�ֶΡ���ͨ�����ܵ������帨��̼Դ���ѽ⣬�Ӷ���Ч����ʯīϩ����������¶ȣ���400�C600�������¼������ʯīϩ������[60-63]��
��֮ǰ����״̬���ڲ�����������ʯīϩ��ͬ��PECVD���õ��ǵ�ѹCVDϵͳ������Ʒ�����η�����һ����������Ƶ�������巢��װ�ã���̼Դ�ѽ�Ϊ�����ߡ���Ӧ����ǿ��̼���֣�ʵ��ʯīϩ�ĵ���������PECVD��ϵ��ʯīϩ����������ͼ5(a)��ʾ���������ͨ����CVDϵͳ�������ڵ������帨�������µ��ѽ�������ף�����CHx<4��̼ԭ�ӵȻ���̼���֣�����̼��Ƭ���������������棬��ʼʯīϩ�ijɺ˺��������̡������ڵ�������������̼Դ�ѽ�ĽϿ죬PECVDʯīϩ�������ٶȱȸ�����CVD������Ҫ�졣�����������¶Ƚϵͣ���������������Ļ���̼���ֵ�Ǩ���˶��ܵ���һ�������ƣ����PECVD������ʯīϩȱ�ݸ��࣬�ᾧ����Ҳ���
��ͼ5(b)��ʾ������ͨ��PECVD�������ڸ��ֲ�ͬ�IJ������������ʯīϩ��������������ͨ�ײ�������ɫ������ʯӢ��������貣��������ʯ���������������������ȣ���Ҳ�ǵ�һ�ζ�ʯīϩ�ڸ��������õ���Ʒ����������ĵ������������е�ϵͳ�о������ڲ�ͬ�����������̼��Ƭ��������Ǩ��������ͬ������ʯīϩ�ڸ��ֲ�����������������в����ͼ5(c)��ʾ�����̲�������������ʯīϩƷ�ʽ���������Ҫ�á�
|
|
|
ͼ4 ʯīϩ������̬�����������������[43] |
|
(a�Cd) morphology evolutions of as-grown samples experiencing different CVD synthetic conditions (The detailed conditions are: (a) 150 cm3�� min�C1Ar/20 cm3�� min�C1H2/8 cm3�� min�C1CH4at 970 ��C for 1 h�� (b) 150 cm3�� min�C1Ar/15 cm3�� min�C1H2/5 cm3�� min�C1CH4at 1000 ��C for 2 h�� (c) 150 cm3�� min�C1Ar/15 cm3�� min�C1H2/6 cm3�� min�C1CH4at 1020 ��C for 2 h�� (d) 150 cm3�� min�C1Ar/15 cm3�� min�C1H2/15 cm3�� min�C1CH4at 1020 ��C for 4 h), as characterized by SEM micrographs and OM images (as insets)�� (e�Cf) AFM height image of the transferred (e) graphene disks and (f) continuous graphene films�� (g) Raman spectra of as-grown graphene films with different layers and bare glass substrates. (h�Ci) SEM images of graphene disks grown at (h) 1000 ��C and (i) 1050 ��C�� (j�Ck) SEM images of graphene disks grown with H2flow rate of (j) 2 cm3�� min�C1and (k) 150 cm3�� min�C1 |
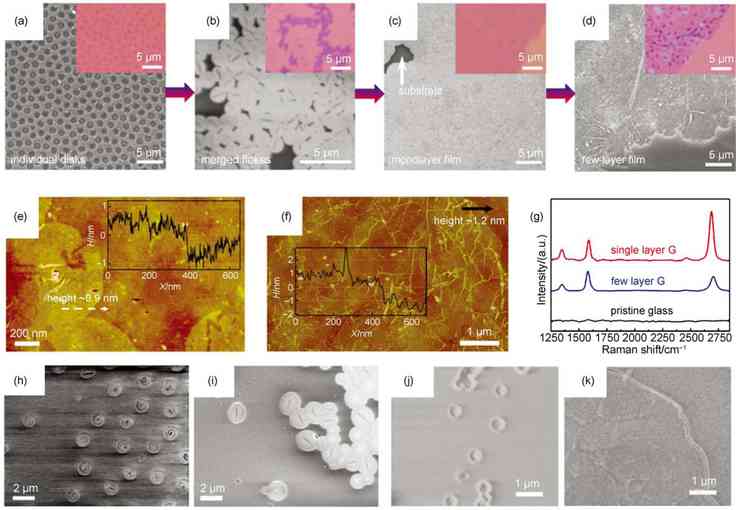
��PECVD����ʯīϩ�����У�̼ԴŨ�ȶ����������Ӱ��ܴ���ͼ5(d)��ʾ����̼ԴŨ�Ⱥ�Сʱ�������ӳ�����ʱ��Ҳ������������ʯīϩ��������̼ԴŨ�ȿ���ʹʯīϩ�ĺ���������ӣ�������Ϊ�и���ļ��鱻�ѽ��������̼���ֲ��뵽ʯīϩ���������̣��ӿ��������ٶȡ����������ڼ���Ũ�Ⱥ���ʱ�ӳ�����ʱ��Ҳ�ܹ���Ч������ʯīϩ��ȡ�����ʯīϩ��ȵIJ�ͬ��������ʯīϩ���������ԵIJ����ͼ5(e)��ʾ����̼ԴŨ������ʱ��ʯīϩ�ĵ�������������������һ���أ����ǿ���ͨ����������������ʵ�ֶ�ʯīϩ��ѧ���Ժ͵����Եĵ��ڣ���ͼ5(f)��ʾ������Ӧ�ö���������Ժ͵����Ե�ʵ��Ҫ�����ı����������������������������������Ҫ�����⣬����ڻ�ԭ����ʯīϩ��ѧ����ʯīϩ�õ�ʯīϩ��Ĥ������ֱ��������ʽ�õ���ʯīϩ��������ͬ��ѧ�����¾��и��õĵ����ԡ�
|
|
|
ͼ5 �������帨��ʯīϩ�ڹ�̬��������ĵ���ֱ������[42] |
|
(a) schematic of graphene growth on glass substrates by plasma enhanced chemical vapor deposition (PECVD)�� (b) photograph of the graphene grown on various types of glasses�� (c) Raman spectra of graphene directly grown on different glass substrates by PECVD under identical conditions�� (d)Raman spectroscopy characterizations of graphene on white float glass by PECVD with different growth parameters�� (e) sheet resistance versus precursor concentration during synthesis for as-grown graphene/white float glass samples. (f) sheet resistance and UV-Vis transmittance spectra of the PECVD graphene directly grown on white float glass�� (g) SEM image of directly grown PECVD vertically oriented graphene films on white float glass�� (h) OM image of the transferred graphene films onto the SiO2/Si substrate�� (i) contact angle versus optical transmittance for graphene/white float glass samples fabricated by direct PECVD. The inset is the demonstration of the hydrophobic and hydrophilic nature of a patterned graphene/glass (the left part) and bare glass surface. |
��ʵ���У����ڵ������巢���������ĵ糡�����벣�������ഹֱ[63,64]��������ʯīϩ������ͼ5(g)��ʾ������״ֱ���ṹ����ͼ5(h)��ʾ����Щ������״ֱ��������ʯīϩ���кܺõ������Ժ;����ԣ�ͬʱ���������õĵ����������Ժͱȶ�άʯīϩ��ö�ıȱ����������ֱ��ʯīϩ��Ĥ�Ǵ�������ƽ̨��������Ⱦ������̫���ܵ�غ���ȡ����⣬���������渲�������ֶ��ص�ʯīϩ��������Իᷢ������ĸı䡣��ͼ5(i)��ʾ���������ͨ����10��C17��ĽӴ��ǣ���ѧ����Ϊ89%��ʯīϩ�����Ӵ��ǿɴ�95�㣬��������ˮ���ܿ�����ʯīϩ��ȵ����Ӷ���һ�����Ӵ������ɴ�143�㣬Զ������ͨ��άʯīϩ����������״ֱ��������ʯīϩ��Ĥ����������õ���ˮ���ܣ�����������صı�����ò�Լ�ʯīϩ�������õ���ˮ���ܣ�����ڷ�չ��ܡ��ͳɱ��������Ѻõ�����ര���Լ�����ˮ�ռ�����dz�������
�ڴ�֮ǰ���о���Ա����PECVD������ʯīϩ�ĵ�����������������о���������һ��ΪSiO2�Ⱦ�Ե����[61,63]�����ڲ�������������о����١��Ź����о���[61]���о�ʯīϩ�ھ�Ե���ĵ�������ʱ��ѡ����ʯӢ������ʯ������Ϊ�������ף�������ʯīϩ��ȴ�ԼΪ2�C3�㣬�����ͼ6(a)��ʾ��ͨ��ͼ6(b)��ʾ�����������Կ��������ڹ��ɱ�Ĥ��ʯīϩ�����ߴ�ֻ�м�ʮ���ף�ʹ��D��ϸߣ��������ǵ����������һ�¡����⣬����ͨ����������ʱ�����ı�ʯīϩ�Ĺ�ѧ���Ժ͵����ԣ������ͼ6(c)��ʾ��ʯīϩ��Ĥ�Ĺ�ѧ����Ϊ92%��85%ʱ���������ֱ�Ϊ40��7 k���C1��Chiu�о���62���õ��ӻ�������������帨����ѧ�������(ECR-PECVD)�ķ���������ϩ��Ϊ̼Դ����ʯӢ���������һ����ʯīϩ�������ɵı�Ĥ�������ͼ6(d)��6(e)��ʾ��ͨ�����ַ����õ���ʯīϩ��Ĥ������ȫ������ʯīϩ��Ĥ�ϴ���ֱ��Ϊ1�C10 nm��С����ͼ6(f)�Ա�����ͬ�����²���ECR-PECVD��ͭ����ʯӢ��������ʯīϩ�Ľ���������������п��Է��֣�����ʯӢ�������Ĵ������Լ��ߵ�Ǩ�����ݣ�����ʯӢ����������ʯīϩ�����dz�С��G����D'�巢���غϣ�2D��Ҳ���Ե���G�塣
|
|
|
ͼ6 �������帨��ʯīϩ��ʯӢ����ĵ�������[61,62] |
|
(a) optical image of a 4-inch wafer scale nanographene film by PECVD�� (b) Raman spectra of nanographene films grown on various substrates��(c) transmittance spectra of samples with different sheet resistance�� (d) direct growth of nanographene on quartz by electron cyclotron resonance(ECR)-CVD. Thickness of nanographene films can be controlled by growth time�� (e) SEM image of nanographene on a quartz plate. The bright area corresponds to the bare substrate made by a scratch�� (f) Raman spectra of ECR-CVD graphene grown on copper (upper) and of ECR-CVD nanographene grown on quartz |
Ŀǰ������PECVD�����ڲ����������������ʯīϩ��������ߴ��С��ȱ�ݺܶ࣬��Ȼ�кܴ�������ռ䣬��ͨ���Ż������������������;������������ʯīϩ������Ʒ�ʡ�
5
ʯīϩ������Ӧ��̽��
һ���²����Ƿ�ɹ����ںܴ�̶���ȡ�������ܷ��ҵ��Լ���Ӧ������ͨ�����Ƿ�չ��ֱ������������ʯīϩ��������һ���Ʊ���ɣ�Ϊ���ģ�����������˿��ܣ���������е���Ҫ��ʯīϩ�����ĸ���Ӧ�ý���̽����Ϊ�ˣ�������ʯīϩ����Ϊԭ���ϣ������������硢���ȡ���ˮ�Լ��������ݵ����ԣ�̽����ʯīϩ�����ڵ��±�ɫ���ڡ������Ӵ���ϸ�������Լ�����ĸ������Ӧ�á�
|
|
|
ͼ7 ʯīϩ����Ӧ��̽��[41,43] |
|
(a) structural schematic of a directly grown graphene glass heater�� (b) cycling performance of the thermochromic display made with graphene/quartz glass (4 cm �� 6 cm�� T550= 85.7%) under an input voltage of 20 V, showing a stable thermochromic behavior over a hundred runs�� the inset presents photographs of the display before and after voltage was applied�� (c) fog removal time and defogger resistance as a function of input voltage�� (d) the defogging behaviors of the DG-based and TG-based defoggers before and after 2 min of ultrasonication treatment�� (e) optical image of 3T3 cells cultured on graphene glasses for 72 h. Inset shows the fluorescent image of cytoskeleton (red) of 3T3 cells cultured on graphene glasses. scale bar: 50 ��m�� (f) statistical histogram of cell proliferation displaying good biocompatibility of graphene glasses�� (g) schematic of graphene glass based photocatalytic plates for degradation of dye wastewater in a recyclable manner�� (h) comparison of the degradation efficiencies of RhB-contained wastewater by using chemically coated BiOF/graphene glass (red), physically coated BiOF/graphene glass (blue), physically coated BiOF/pure glass(pink), pure graphene glass (green), and pure glass (black) as photocatalysts under natural sunlight irradiation�� the inset displays a photograph of prototype reactors after photodegradation by employing the marked catalysts�� (i) cycling photodegradation of RhB-contained solution using chemically coated BiOF/graphene glass plates |
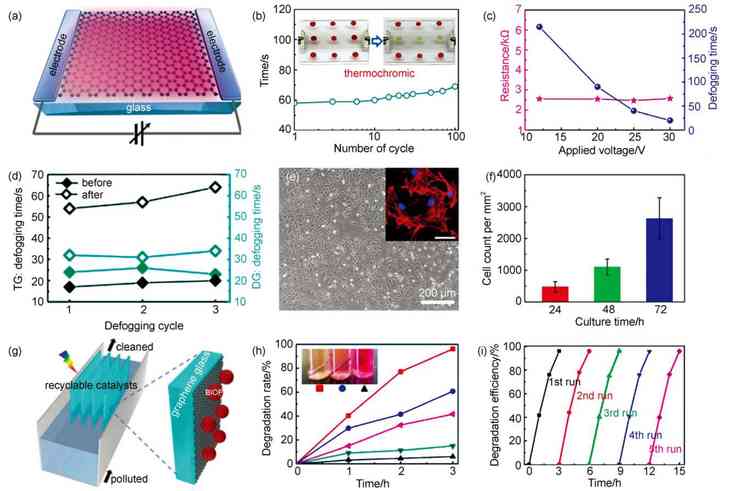
���±�ɫ���ڿ���ͨ��ʩ�ӵ�ѹ�Դ��ڵ����Ժ���ɫ���е��ڣ������ܽ��������ܼҾ��а�����Ҫ��ɫ���ڸ������͵ĵ��±�ɫ�����У�������������Ƿdz���Ҫ����ɲ��֣�ʯīϩ���������˲��������Ժ�ʯīϩ�ĵ��絼�����ܣ����з�չ���±�ɫ���ڵ�DZ��������ʯīϩ���������ĵ������У�ʩ�ӵ�ѹʱ���������(�������)����һ��ߴ�Ϊ1 cm �� 2 cmʯīϩ������ʩ��20 V�ĵ�ѹ��ʯīϩ������¶ȱ�ɴﵽ(43.5 �� 1.3)�����������¶ȷֲ����ȣ�����������ɫ���ڵ�Ҫ����ͼ7(a��b)��ʾ�����������˻���ʯīϩ�����ĵ��±�ɫ���ڣ���4 cm �� 6 cmʯӢ�����ʯīϩ��ʴ������1.5 cm �� 6 cm����������ÿ�������϶�Ϳ�����±�ɫ��(��ɫת���¶�Ϊ39��)�����м�ʯīϩ��������ʩ��20 V��ѹ�����±�ɫ����60 s���ɰ���ɫ��Ϊdz��ɫ�������������ϵı�ɫ����ɫ�ޱ仯���жϵ�Դ���ɫ���ָ�����ɫ������ʯīϩ���������ĵ��±�ɫ���ھ��кܺõ��ظ��ԡ�
�����Ӵ��ǻ���ʯīϩ���������絼�����ʵ���һ����ҪӦ�á�����ˮ���IJ���Դ�ڴ�������ˮ�鸲���ڲ����ı��棬��˳����ķ����Ӵ�һ��ͨ��������ˮ����������ˮ�����߱�����ȵķ�ʽ����ֹˮ�����Ӵ�����ĸ��ǡ�����ʯīϩ�����ĵ����Ժ���ˮ�ԣ�ʹ��ʯīϩ����ͬʱ��߱�����ˮ���ͱ�����ȵ��������ȿ���ͨ����ˮ�Դ��ֹ�����γɣ����ܹ����������ͨ������Ƚ��г�������ˣ�ʯīϩ�����Ӵ�ͬʱ���б���������������������һ������ķ����Ӵ�ԭ���ϡ����Ƕ�ʯīϩ����(2 cm �� 2 cm)�ij������������˲��ԣ������ͼ7(c)��ʾ������ʯīϩ��������ʩ�Ӳ�ͬ�ĵ�ѹʱ������ʱ�����ѹ��������������̣������˵�ѹΪ30 Vʱ�����������ˮ����21 s�ڱ���ȫ�����������������ʯīϩ�ij�������[65��66]����������������ʯīϩ�����ĵ��������Ա仯��������Ϊʯīϩ�벣��֮��ĽӴ��dz����ܣ�ˮ��������ʯīϩ�Ͳ������м���棬�Ե���Ӱ���С[55]�����ǻ��Ա���CVDֱ������(DG)��ת��(TG)���ַ����Ʊ���ʯīϩ�����ij��������������ͼ7(d)��ʾ����ʼʱ����ʯīϩ�����ij��������൱��������2 min����������ֱ�������õ���ʯīϩ�����������ܱ仯����ת�Ƶõ���ʯīϩ������������ȴ����½���˵��ֱ�������ķ����ܹ����������ʯīϩ�벣��֮�����������ʹʯīϩ���������ȶ���
����ʯīϩ�������õ����������ԣ�������ϸ����ֳ������ʯīϩ�����Ƴɵ�������Ȳ�������������Ϊһ������ʵ����Ʒ�����ǽ�ʯīϩ������ΪNIH-3T3ϸ��[67]������ý�飬����DAPIȾɫ�����о�ϸ����ʯīϩ�����������������ֳ�����ͼ7(e)չʾ��ϸ��������72 h��Ľ������ʯīϩ����������ֳ����õ���ֳ��Ϊ������Ⱦɫ�������������ع۲쵽ϸ��������������ò��Ǽܣ�˵��ʯīϩ�����ϸ���ṹ����Ӱ�졣ͼ7(f)ͨ��ͳ��ϸ����24��48��72 h����Ŀ��չʾ��ʯīϩ�������õ����������ԡ�
���⣬�ڲ�������ֱ�������õ���ʯīϩ�����Ժܺã���һ�ֹ����������塣BiOF��һ�ֳ����Ŀɼ��⼤���Ĺ��������ͼ7(g)��ʾ������ͨ����ѧ�����������BiOF���ص�CVDֱ�������õ���ʯīϩ�������棬������ˮ��Ⱦ�ϵĹ�����⡣������BΪ����������BiOF��ʯīϩ�������ֳ��ܺõĹ�����ԣ���ͼ7(h)��ʾ������Ȼ������3 h������B�Ľ����ʸߴ�96.2%��Զ����ֱ���ڲ������渺��BiOF�Ľ���Ч����BiOF��ʯīϩ�����ϵĴ�Ч��֮�����������ڲ���������Ϊʯīϩ�Ĵ��ڽ����˵���-��Ѩ�ĸ�������[68,69]��ͼ7(i)չʾ�����ָ��Ϲ�������ѭ��ʹ�����������ʹ�ú�����Ч��û�����Խ��ͣ����ֳ����õ��ȶ��ԡ���BiOF֮�⣬���ǽ�����ʯīϩ�����Ĵ�������չ������������ϵ����BiVO4��Bi2WO6�����ܱ��ֳ�����Ĺ�����ܡ���ˣ�ʯīϩ������һ�־��������ԵĹ�����塣
����ʹ�õ��ձ��Ժ�ʯīϩ���ܵĶ����Ծ�����ʯīϩ����Ӧ�õĹ㷺�ԣ���������չʾ������ʵ����ʯīϩ�����������ճ������е���;���кܶ࣬��Ҫ���ǽ�һ��ȥ�����̽����
6
�ܽ���չ��
����ϵͳ��չʾ������CVD�����ڲ�������ֱ������ʯīϩ����ع�������Բ�ͬ����IJ����Լ�ʵ��Ӧ�õ���Ҫ�����Ƿֱ�չ�����ֲ�ͬ�������������������²��������Dz��ó�ѹCVD��ϵ��ͨ���Ż����������������²����������������Ʒ�ʽϸߵĵ���Ͷ��ʯīϩ[41]��ʯīϩ�ĺ�ȿ���ͨ�������¶ȡ�����ʱ���Լ�̼ԴŨ�Ƚ�����Ч���ء������ͨ������ͬ�����ó�ѹCVD��ϵ�������Եط�չ������̬���������ʯīϩ��������[43]����������ʯīϩ���зֲ����ȡ��ߴ��һ���ص㡣����ڹ�̬������ʯīϩ������̬��������������ٶȿ�(Լ10��)����������¶Ⱥ�̼ԴŨ�ȶ�������Ч��������̬��������ʯīϩ�ĸ��Ƕȡ������ѳ��͵Ĺ�̬������Ϊ���������κ��������ʣ����Dz���PECVD�����������������ʯīϩ[42]����ͨ���Ե������巢�����ĵ��ڣ�������������״ֱ����ʯīϩ��Ĥ����Ĥ�ĺ�ȿ���ͨ������̼ԴŨ�Ⱥ�����ʱ�������ơ�
�����ͨ��Һ��ͿĤ��ת�Ʒ����Ʊ���ʯīϩ��������������ֱ��������ʯīϩ���ж��ص����ơ�һ���棬����ʯīϩ�벣��֮��Ӵ����ܣ��������ǿ��������࣬���кܺõ��ȶ��ԡ���һ���棬Һ��ͿĤ��ת�Ƶõ���ʯīϩ��Ĥ���������������Ⱦ���ⶼ���ʯīϩ������ɼ�����ƻ�����ֱ���������ԴӸ�Դ�ϱ�����Щ���⡣���ͬʱ������ֱ���ڲ�������������һ���Ʊ��������������߱�ʵ��ʯīϩ������ģ�������������������ƶ�ʯīϩ��������Ӧ�þ�����Ҫ���á�
ʯīϩ���������е������硢���ȡ�������ˮ�Լ����������Ե��ص㣬ʹ��ʯīϩ�������й㷺��Ӧ��DZ��������ͨ����չ����ʯīϩ�����ĵ��±�ɫ���ڡ������Ӵ���ϸ�����������Լ�������壬չʾ��δ��ʯīϩ�����ڽ�������������Դ������������ҽҩ����������Ӧ��ǰ������ˣ�ʯīϩ������ʵ��ʯīϩ��ҵ��Ӧ�õ�һ����ʵ���еĵ�·��
ʯīϩ���������ֳ����������ܼ���Ӧ�ü�ֵֵ�����Ǽ�����ע�������о���Ŀǰ��ʯīϩ������������Ӧ���о����ڿ�ѧ�ͼ��������ɴ����ڶ�����û�н���������һ����߲�������ʯīϩ�����������������¶ȣ���������ʱ�䣬ʵ��ʯīϩ�����Ĺ�ҵ����������չ�µ��м�ֵ��Ӧ�õȡ�����ڹ����й�������˵��������ս���ǻ��������������о������룬��Щ�����ջ�����ʯīϩ��������Ӧ�����ճ������еķ������档
�����
[1]Novoselov, K. S.; Geim, A. K.; Morozov, S. V.; Jiang, D.;Zhang, Y.; Dubonos, S. V.; Grigorieva, I. V.; Firsov, A. A. Science 2004, 306, 666. doi: 10.1126/science.1102896
[2]Novoselov, K. S.; Fal'ko, V. I.; Colombo, L.; Gellert, P. R.;Schwab, M. G.; Kim, K. Nature 2012, 490, 192. doi: 10.1038/nature11458
[3]Yan, K.; Fu, L.; Peng, H.; Liu, Z. Accounts Chem. Res. 2013, 46, 2263. doi: 10.1021/ar400057n
[4]Bonaccorso, F.; Sun, Z.; Hasan, T.; Ferrari, A. C. Nature Photon. 2010, 4, 611. doi: 10.1038/nphoton.2010.186
[5]Geim, A. K.; Novoselov, K. S. Nature Mater. 2007, 6, 183. doi: 10.1038/nmat1849
[6]Geim, A. K. Science 2009, 324, 1530. doi: 10.1126/science.1158877
[7]Nair, R. R.; Blake, P.; Grigorenko, A. N.; Novoselov, K. S.;Booth, T. J.; Stauber, T.; Peres, N. M. R.; Geim, A. K. Science 2008, 320, 1308. doi: 10.1126/science.1156965
[8]Geim, A. K. Rev. Mod. Phys. 2011, 83, 851. doi: 10.1103/RevModPhys.83.851
[9]Novoselov, K. S. Rev. Mod. Phys. 2011, 83, 837. doi: 10.1103/RevModPhys.83.837
[10]Lee, C.; Wei, X.; Kysar, J. W.; Hone, J. Science 2008, 321, 385. doi: 10.1126/science.1157996
[11]Du, X.; Skachko, I.; Barker, A.; Andrei, E. Y. Nature Nanotech. 2008, 3, 491. doi: 10.1038/nnano.2008.199
[12]Seol, J. H.; Jo, I.; Moore, A. L.; Lindsay, L.; Aitken, Z. H.;Pettes, M. T.; Li, X.; Yao, Z.; Huang, R.; Broido, D.; Mingo, N.;Ruoff, R. S.; Shi, L. Science 2010, 328, 213. doi: 10.1126/science.1184014
[13]Bae, S.; Kim, H.; Lee, Y.; Xu, X.; Park, J. S.; Zheng, Y.;Balakrishnan, J.; Lei, T.; Kim, H. R.; Song, Y. I.; Kim, Y. J.;Kim, K. S.; Ozyilmaz, B.; Ahn, J. H.; Hong, B. H.; Iijima, S. Nature Nanotech. 2010, 5, 574.
[14]Liu, C.; Yu, Z.; Neff, D.; Zhamu, A.; Jang, B. Z. Nano Lett. 2010, 10, 4863. doi: 10.1021/nl102661q
[15]Lin, Y. M.; Valdes-Garcia, A.; Han, S. J.; Farmer, D. B.; Meric, I.; Sun, Y.; Wu, Y.; Dimitrakopoulos, C.; Grill, A.; Avouris, P.;Jenkins, K. A. Science 2011, 332, 1294. doi: 10.1126/science.1204428
[16]Xia, F.; Mueller, T.; Lin, Y. M.; Valdes-Garcia, A.; Avouris, P. Nature Nanotech. 2009, 4, 839. doi: 10.1038/nnano.2009.292
[17]Park, J.; Ahn, Y. H.; Ruiz-Vargas, C. Nano Lett. 2009, 9, 1742. doi: 10.1021/nl8029493
[18]Koppens, F. H. L.; Mueller, T.; Avouris, P.; Ferrari, A. C.;Vitiello, M. S.; Polini, M. Nature Nanotech. 2014, 9, 780.
[19]Yoo, E.; Kim, J.; Hosono, E.; Zhou, H. S.; Kudo, T.; Honma, I. Nano Lett. 2008, 8, 2277. doi: 10.1021/nl800957b
[20]Xu, M.; Fujita, D.; Hanagata, N. Small 2009, 5, 2638. doi: 10.1002/smll.v5:23
[21]Garaj, S.; Hubbard, W.; Reina, A.; Kong, J.; Branton, D.;Golovchenko, J. A. Nature 2010, 467, 190. doi: 10.1038/nature09379
[22]Xing, F.; Liu, Z. B.; Deng, Z. C.; Kong, X. T.; Yan, X. Q.; Chen, X. D.; Ye, Q.; Zhang, C. P.; Chen, Y. S.; Tian, J. G. Sci. Rep. 2012, 2, 908. doi: 10.1038/srep00908
[23]Xing, F.; Meng, G. X.; Zhang, Q.; Pan, L. T.; Wang, P.; Liu, Z. B.; Jiang, W. S.; Chen, Y.; Tian, J. G. Nano Lett. 2014, 14, 3563. doi: 10.1021/nl5012036
[24]Xu, W.; Ling, X.; Xiao, J.; Dresselhaus, M. S.; Kong, J.; Xu, H.;Liu, Z.; Zhang, J. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 9281. doi: 10.1073/pnas.1205478109
[25]Paton, K. R.; Varrla, E.; Backes, C.; Smith, R. J.; Khan, U.;O'Neill, A.; Boland, C.; Lotya, M.; Istrate, O. M.; King, P.;Higgins, T.; Barwich, S.; May, P.; Puczkarski, P.; Ahmed, I.;Moebius, M.; Pettersson, H.; Long, E.; Coelho, J.; O'Brien, S. E.;McGuire, E. K.; Sanchez, B. M.; Duesberg, G. S.; McEvoy, N.;Pennycook, T. J.; Downing, C.; Crossley, A.; Nicolosi, V.;Coleman, J. N. Nature Mater. 2014, 13, 624. doi: 10.1038/nmat3944
[26]Li, X.; Zhang, G.; Bai, X.; Sun, X.; Wang, X.; Wang, E.; Dai, H. Nature Nanotech. 2008, 3, 538. doi: 10.1038/nnano.2008.210
[27]Dai, B.; Fu, L.; Zou, Z.; Wang, M.; Xu, H.; Wang, S.; Liu, Z. Nature Commun. 2011, 2, 522. doi: 10.1038/ncomms1539
[28]Li, X.; Cai, W.; An, J.; Kim, S.; Nah, J.; Yang, D.; Piner, R.;Velamakanni, A.; Jung, I.; Tutuc, E.; Banerjee, S. K.; Colombo, L.; Ruoff, R. S. Science 2009, 324, 1312. doi: 10.1126/science.1171245
[29]Kim, K. S.; Zhao, Y.; Jang, H.; Lee, S. Y.; Kim, J. M.; Kim, K. S.; Ahn, J. H.; Kim, P.; Choi, J. Y.; Hong, B. H. Nature 2009,457, 706. doi: 10.1038/nature07719
[30]Gao, T.; Xie, S.; Gao, Y.; Liu, M.; Chen, Y.; Zhang, Y.; Liu, Z. ACS Nano 2011, 5, 9194. doi: 10.1021/nn203440r
[31]Pan, Y.; Zhang, H.; Shi, D.; Sun, J.; Du, S.; Liu, F.; Gao, H. J. Adv. Mater. 2009, 21, 2777. doi: 10.1002/adma.200800761
[32]Gao, L.; Ren, W.; Xu, H.; Jin, L.; Wang, Z.; Ma, T.; Ma, L. P.;Zhang, Z.; Fu, Q.; Peng, L. M.; Bao, X.; Cheng, H. M. Nature Commun. 2012, 3, 699. doi: 10.1038/ncomms1702
[33]Coraux, J.; N'Diaye, A. T.; Busse, C.; Michely, T. Nano Lett. 2008, 8, 565. doi: 10.1021/nl0728874
[34]Chae, S. J.; Guenes, F.; Kim, K. K.; Kim, E. S.; Han, G. H.; Kim, S. M.; Shin, H. J.; Yoon, S. M.; Choi, J. Y.; Park, M. H.; Yang, C. W.; Pribat, D.; Lee, Y. H. Adv. Mater. 2009, 21, 2328. doi: 10.1002/adma.v21:22
[35]Li, X.; Cai, W.; Colombo, L.; Ruoff, R. S. Nano Lett. 2009, 9, 4268. doi: 10.1021/nl902515k
[36]Chen, J. S.; Wu, B.; Liu, Y. Q. Acta Chim. Sin. 2014, 72, 355.�۳¼�˼, �� ��, ������. ��ѧѧ��, 2014, 72, 355.��
[37]Liu, N.; Fu, L.; Dai, B.; Yan, K.; Liu, X.; Zhao, R.; Zhang, Y.;Liu, Z. Nano Lett. 2011, 11, 297. doi: 10.1021/nl103962a
[38]Liu, M.; Zhang, Y.; Chen, Y.; Gao, Y.; Gao, T.; Ma, D.; Ji, Q.;Zhang, Y.; Li, C.; Liu, Z. ACS Nano 2012, 6, 10581.
[39]Zhang, C. H.; Fu, L.; Zhang, Y. F.; Liu, Z. F. Acta Chim. Sin. 2013, 71, 308. ���ų���, �� ��, ����, ���ҷ�. ��ѧѧ��, 2013, 71, 308.�� doi: 10.6023/A13010023
[40]Zou, Z. Y.; Dai, B. Y.; Liu, Z. F. Sci. Sin. Chim. 2012, 42, 1. ����־��, ������, ���ҷ�. �й���ѧ: ��ѧ, 2012, 42, 1.��
[41]Sun, J.; Chen, Y.; Priydarshi, M. K.; Chen, Z.; Bachmatiuk, A.;Zou, Z.; Chen, Z.; Song, X.; Gao, Y.; Ruemmeli, M. H.; Zhang, Y.; Liu, Z. Nano Lett. 2015, 15, 5846. doi: 10.1021/acs.nanolett.5b01936
[42]Sun, J.; Chen, Y.; Cai, X.; Ma, B.; Chen, Z.; Priydarshi, M. K.;Chen, K.; Gao, T.; Song, X.; Ji, Q.; Guo, X.; Zou, D.; Zhang, Y.;Liu, Z. Nano Res. doi: 10.1007/s12274-015-0849-0
[43]Chen, Y.; Sun, J.; Gao, J.; Du, F.; Han, Q.; Nie, Y.; Chen, Z.;Bachmatiuk, A.; Priydarshi, M. K.; Ma, D.; Song, X.; Wu, X.;Xiong, C.; Ruemmeli, M. H.; Ding, F.; Zhang, Y.; Liu, Z. Adv. Mater. doi: 10.1002/adma.201504229
[44]Bhaviripudi, S.; Jia, X.; Dresselhaus, M. S.; Kong, J. Nano Lett. 2010, 10, 4128. doi: 10.1021/nl102355e
[45]Zhao, P.; Kumamoto, A.; Kim, S.; Chen, X.; Hou, B.; Chiashi, S.; Einarsson, E.; Ikuhara, Y.; Maruyama, S. J. Phys. Chem. C 2013, 117, 10755. doi: 10.1021/jp400996s
[46]Zhao, P.; Kim, S.; Chen, X.; Einarsson, E.; Wang, M.; Song, Y.;Wang, H.; Chiashi, S.; Xiang, R.; Maruyama, S. ACS Nano 2014,8, 11631. doi: 10.1021/nn5049188
[47]Chen, J.; Guo, Y.; Jiang, L.; Xu, Z.; Huang, L.; Xue, Y.; Geng, D.; Wu, B.; Hu, W.; Yu, G.; Liu, Y. Adv. Mater. 2014, 26, 1348. doi: 10.1002/adma.201304872
[48]Wei, D.; Lu, Y.; Han, C.; Niu, T.; Chen, W.; Wee, A. T. S. Angew. Chem. Int. Edit. 2013, 52, 14121. doi: 10.1002/anie.201306086
[49]Hwang, J.; Kim, M.; Campbell, D.; Alsalman, H. A.; Kwak, J. Y.; Shivaraman, S.; Woll, A. R.; Singh, A. K.; Hennig, R. G.;Gorantla, S.; Ruemmeli, M. H.; Spencer, M. G. ACS Nano 2013,7, 385. doi: 10.1021/nn305486x
[50]Chen, J.; Wen, Y.; Guo, Y.; Wu, B.; Huang, L.; Xue, Y.; Geng, D.; Wang, D.; Yu, G.; Liu, Y. J. Am. Chem. Soc. 2011, 133, 17548. doi: 10.1021/ja2063633
[51]Sun, J.; Gao, T.; Song, X.; Zhao, Y.; Lin, Y.; Wang, H.; Ma, D.;Chen, Y.; Xiang, W.; Wing, J.; Zhang, Y.; Liu, Z. J. Am. Chem. Soc. 2014, 136, 6574. doi: 10.1021/ja5022602
[52]Teng, P. Y.; Lu, C. C.; Akiyama-Hasegawa, K.; Lin, Y. C.; Yeh, C. H.; Suenaga, K.; Chiu, P. W. Nano Lett. 2012, 12, 1379. doi: 10.1021/nl204024k
[53]Kim, H.; Song, I.; Park, C.; Son, M.; Hong, M.; Kim, Y.; Kim, J. S.; Shin, H. J.; Baik, J.; Choi, H. C. ACS Nano 2013, 7, 6575. doi: 10.1021/nn402847w
[54]Dong, X.; Wang, P.; Fang, W.; Su, C. Y.; Chen, Y. H.; Li, L. J.;Huang, W.; Chen, P. Carbon 2011, 49, 3672. doi: 10.1016/j.carbon.2011.04.069
[55]Tan, L.; Zeng, M.; Wu, Q.; Chen, L.; Wang, J.; Zhang, T.;Eckert, J.; Ruemmeli, M. H.; Fu, L. Small 2015, 11, 1840. doi: 10.1002/smll.201402427
[56]Geng, D.; Wu, B.; Guo, Y.; Huang, L.; Xue, Y.; Chen, J.; Yu, G.;Jiang, L.; Hu, W.; Liu, Y. Proc. Natl. Acad. Sci. U. S. A. 2012,109, 7992.
[57]Zeng, M.; Tan, L.; Wang, J.; Chen, L.; Ruemmeli, M. H.; Fu, L. Chem. Mater. 2014, 26, 3637. doi: 10.1021/cm501571h
[58]Wang, J.; Zeng, M.; Tan, L.; Dai, B.; Deng, Y.; R��mmeli, M.;Xu, H.; Li, Z.; Wang, S.; Peng, L.; Eckert, J.; Fu, L. Sci. Rep. 2013, 3, 2670.
[59]Guqiao, D.; Yun, Z.; Shumin, W.; Qian, G.; Lei, S.; Tianru, W.;Xiaoming, X.; Mianheng, J. Carbon 2013, 53, 321. doi: 10.1016/j.carbon.2012.11.018
[60]Munoz, R.; Gomez-Aleixandre, C. J. Phys. D: Appl. Phys. 2014,47.
[61]Zhang, L.; Shi, Z.; Wang, Y.; Yang, R.; Shi, D.; Zhang, G. NanoRes. 2011, 4, 315. doi: 10.1007/s12274-010-0086-5
[62]Medina, H.; Lin, Y. C.; Jin, C.; Lu, C. C.; Yeh, C. H.; Huang, K. P.; Suenaga, K.; Robertson, J.; Chiu, P. W. Adv. Funct. Mater. 2012, 22, 2123. doi: 10.1002/adfm.201102423
[63]Yang, C.; Bi, H.; Wan, D.; Huang, F.; Xie, X.; Jiang, M. J. Mater. Chem. A 2013, 1, 770. doi: 10.1039/C2TA00234E
[64]Zhu, M. Y.; Outlaw, R. A.; Bagge-Hansen, M.; Chen, H. J.;Manos, D. M. Carbon 2011, 49, 2526. doi: 10.1016/j.carbon.2011.02.024
[65]Wang, J.; Fang, Z.; Zhu, H.; Gao, B.; Garner, S.; Cimo, P.; Barcikowski, Z.; Mignerey, A.; Hu, L. Thin Solid Films 2014,556, 13. doi: 10.1016/j.tsf.2013.12.060
[66]Sui, D.; Huang, Y.; Huang, L.; Liang, J.; Ma, Y.; Chen, Y. Small 2011, 7, 3186. doi: 10.1002/smll.v7.22
[67]Ryoo, S. R.; Kim, Y. K.; Kim, M. H.; Min, D. H. ACS Nano 2010, 4, 6587. doi: 10.1021/nn1018279
[68]Huang, X.; Qi, X.; Boey, F.; Zhang, H. Chem. Soc. Rev. 2012,41, 666. doi: 10.1039/C1CS15078B
[69]Chang, H.; Wu, H. Energy Environ. Sci. 2013, 6, 3483.