光化学反应的从头算研究* yd14510
丁万见 方维海** 北京师范大学化学学院 北京100875
收稿:2007年8月(特约)
*国家自然科学基金项目(No.20472011,20233020,20403003)和国家重点基础研究发展计划(973)项目(No.2004CB719903,2002CB613406)资助
**通讯联系人e-mail:fangwh@bnu.edu.CB
原载:化学进展,2007/10;1449-1459
【摘要】光化学反应是最基本也是最重要的物理化学过程之一,在诸多领域有着广泛的应用。由于计算方法的限制以及光化学反应过程的错综复杂性,光化学反应机理的从头算研究是极具挑战性的国际前沿课题之一。本文综述了近20年来羰基化合物光化学反应机理从头算研究的一些进展,总结了羰基化合物电子激发态的特性和光化学反应过程的规律性,为深入研究光化学反应提供一些有用的信息。
【关键词】光化学 反应机理 从头算研究 CASSCF 电子激发态 羰基化合物
【中图分类号】0641.12+ 1;0644.13 文献标识码:A 文章编号:1005-281X(2007)10-1449-11
光化学反应是最基本也是最重要的物理化学过程之一。它与许多生命过程紧密相关,在环境保护、新材料、新能源和信息技术等领域也发挥着独特的作用。光化学反应一直是极为活跃的化学和物理研究课题,备受理论和实验化学家的关注。光化学反应总要涉及电子激发态,其寿命短,实验上难以观察;光诱导的反应有多种能量可及的途径,且可能在不同的电子态上进行;光物理和光化学反应互相竞争,内转换、系间窜跃和化学反应诸速率共同决定最终产物的状态。与热化学反应相比,光化学反应过程是错综复杂的。除特殊情况外,建立在单参考态基础上的从头算方法,不能用来优化激发态反应途径。多参考态方法,如MR.CI(multi。Reference configuration interaction)方法,可用来描述激发态反应途径。但是多参考态组态相互作用方法耗时长,对计算机硬件要求高,仅可研究小的分子体系。由于光化学反应的复杂性和从头算方法的局限性,光诱导化学过程的从头算研究是一个极具挑战性的理论课题。
尽管如此,经过几十年的努力,人们对于光化学反应的理论研究还是取得了很大的进展。羰基化合物是一类非常重要的化合物,实验和理论工作者对此类化合物的光化学反应进行了系统的研究[1-6]。处于激发态的酮类化合物主要发生两类反应:α-解离(Norrish I)和1,5-氢迁移并继而进行解离或环化反应(Norrish II 或Norrish-Yang)。Norrish I 和Norrish II的反应机理以及动力学行为是理解羰基化合物光化学反应的关键。本文将以脂肪族和芳香族羰基化合物为代表,从理论上探讨它们光解离反应机理的规律性。
1 脂肪族羰基化合物的光解反应
1.1 丙酮及相关的脂肪酮
对脂肪族羰基化合物的研究已经持续了几十年。丙酮(cH3COCH3)作为最简单的脂肪酮,是一个非常经典的例子。一系列理论研究[7-12]使人们对丙酮.S1态的动力学有了深刻的认识。在n-π・ 跃迁的情况下,CH3COCH3(S1)可以通过.S0/S1交叉回到基态,进而在基态发生α键解离;也可以通过S1附近的Franck-Condon区域经由S1/T1到达最低三态T1势能面,α-C―C键断裂在T1态进行。对丙酮而言,α-C―C解离在S1(1nπ・)还是T1 (3nπ・)势能面上进行依赖于激发光波长。图1给出了丙酮光解反应的势能剖面示意图。
|
|
|
图1 丙酮光解势能剖面示意图及相应驻点的几何和电子结构 (键长:X 10 nm;键角:度)、相对基态的能量(eV)[13] |
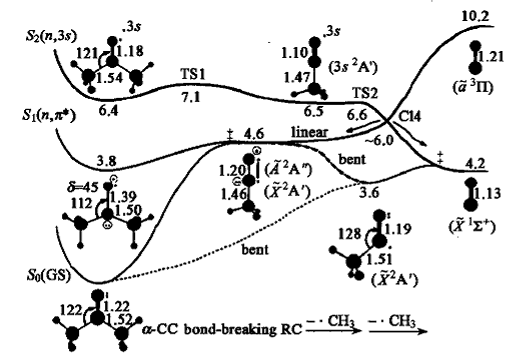
丙酮的第二个吸收带(S0→S2)来源于Rydberg态的电子跃迁(ny→3s),该激发态具有B2对称性。为了重现实验得到的垂直激发光谱,人们进行了一些量子化学计算[14-18]。计算研究发现,在限制C2V对称性的情况下,丙酮的A1(π→π・)势能面与另外两个A1Rydberg态势能面沿着CO伸缩振动坐标交叉。Zewail等的理论和实验研究[13,19]认为,处于S2态的丙酮进行解离有两种不同的反应机理:当体系的内能足以克服67 kJ/mol(16 kcal/mo1)的能垒时,两个C―C键在S2势能面上进行分步解离;而当激发能与S0→S2的带源能量接近时,体系只能通过相对较慢的S1→S2振动相互作用到达S1势能面并进而进行α-C―C解离。
人们应用完全活化空间自洽场(complete active space self-consistent field,CASSCF)和时间相关的密度泛函(TD-DFT)方法,研究了取代基对α-C―C解离的影响[7]。对于甲基取代的脂肪酮,其在S1势能面上的α-C―C解离的能垒表现出引人注意的规律。对于对称取代的酮如RCOR(R=CH3、CH2CH3、CH(CH3)2、C(CH3)3),其α-C―C键的解离能垒是系统地降低的,从CH3COCH3中的76.9 kJ/mol(18.4kcal/mo1)降低到CH3CH2COCH2CH3中的51.4 kJ/mol(12.3 kcal/mo1),进而降低到(CH3)2CHCOCH(CH3)2的30.9 kJ/mol(7.4 kcal/mo1)和(CH3)3CCOC(CH3)3的31.8 kJ/mol(7.6 kcal/mo1)。解离能垒的显著降低预示着α-C―C解离越来越容易发生。
1.2 乙酰氯(CH3COC1)和溴代乙酰氯(BrCH2COC1)
对于非对称取代的羰基化合物,键的选择性解离以及能量分配问题引起了人们的高度兴趣。由于不同键的解离具有可比的指前因子,因此α键的相对强度应该与相应的键解离能垒的相对高低成正比。一般认为[5,6]在低能量光子的激发下,两个α键中较弱的一个优先断裂。但是,很多非烷基取代的羰基化合物的光解实验研究表明,两个α键中较强的一个具有更高的产率,或者两个强度相近的化学键的光解产率明显不同。例如CH3COX(X=C1、Br、I) [20-28]、CH3 COOH[29-31]、BrCH2 COC1[26,27,30-37]等体系的情况。对这类化合物XCOY(X=H、CH3、CH3CH2、CH3CH2CH2、OH、NH2;Y = H、F、OH、C1、CH3、CN、C6H5)的n→π・跃迁下的光解反应进行的系统的理论研究[9,38-55]发现,键的强弱仅仅是影响α键断裂选择性的一个因素,α键断裂的选择性主要取决于解离机理。
C―C和C―C1键的键解离能分别是359.1和342.3 kJ/mol(85.9和81.9 kcal/mo1),两个键的强度相近。早期人们据此推测两个键的解离程度相近。但最近实验观测到,CH3COC1光解时C―Cl键优先解离。图2给出了CH3COC1的两个α键解离的态相关示意图及MR-CI计算的驻点的相对能量。基态Cl原子具有 2P谱项,是三重简并的。而自由基碎片CH3CO具有非简并的 2A’ 基态。当两个基态自由基碎片以Cs 或C1 对称性接近时,它们可以与CH3COC1的6个能量最低的电子态S0、S1、S2、T1、T2和T3绝热相关。脂肪族羰基化合物的紫外激发通常导致分子被激发到S1 ( 1nπ・ )电子态。CH3COC1的α-C―Cl S1解离产生自由基碎片CH3CO(X2A‘)+Cl(2P),由于光解碎片都处于基态,可以预见α-C―Cl键在S1势能面上的解离具有较低的能垒。
以CASSCF优化的平衡结构为基础,MR-CI计算预测α-C―Cl键的S1解离能垒为6.3 kJ/mol(1.5kcal/mo1)。如图2所示,
|
|
|
图2 CH3COC1中两个α键解离的势能剖面示意图 (能量数据来源于MR-CI计算,单位:kcal/mo1) |
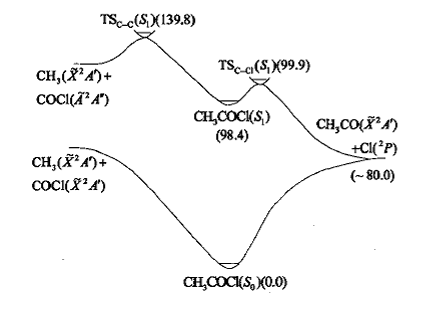
基态CH3 ( X2A’) + COC1( X2A’ )自由基只能与CH3COC1的S0和T1电子态绝热相关,而α-C―C键在S1势能面上的解离只能与处于激发态的自由基绝热相关,因此α-C―C键的S1解离会具有较高的能垒,该能垒在同样的计算水平上被预测为173.1 kJ/mol(41.4 kcal/mo1)。上述的定性态相关分析和定量的MR-CI计算合理地解释了为什么C―C和C―Cl键的强度相当,但是在n→π・跃迁的情况下α-C―C解离不能与α-C―Cl解离相竞争的实验事实。在BrCH2COC1分子中,与羰基相连的有两个不同的α键和一个较弱的β键,因而BrCH2COC1是研究光诱导的键的选择性解离的极好例子。实验[32,33]发现,BrCH2COC1分子在248 nm激光的照射下,较强的C―Cl α键优先于较弱的C―Br β键解离,而且由于n→π・ 跃迁形成的C―Cl α和C―Brβ解离之间的支化率被推测为1.0:0.4; C―C α键的解离也是可以发生的,只是几率非常小。以CASSCF和MR-Cl计算得到的势能面为基础[53,54],结合非绝热过渡态理论和从头算分子动力学计算,n→π・跃迁情况下C―Cl α和C―Br β键解离的选择性问题得到了详细的研究。
研究表明在C―Cl解离的过渡态区域,S1和S2之间的能隙为7 372 cm-1,如图3所示。
|
|
|
图3 BrCH2COC1中C―Cl和C―Br键解离的势能剖面示意图及相应的驻点结构(键长:X 10 nm;键角:度;能量:cm-1 ;MR-CI/cc-pVDZ) [54] |
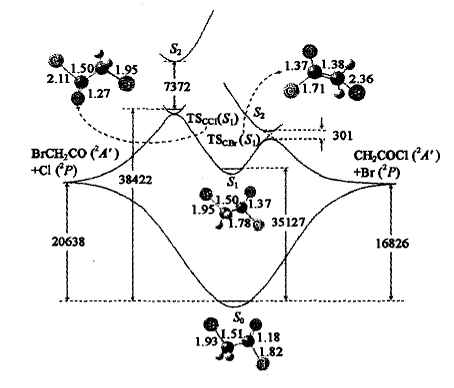
由于S1和S2态之间的避免相互作用非常强,C―Cl键沿S1势能面的解离可以看成是绝热过程。而在C―Br解离的过渡态区域,S1和S2之间的避免作用非常弱(耦合矩阵元为150.5 cm-1 ),因此,C―Br键的S1解离是一个非绝热过程。若所给激发能为482.4 kJ/mol(248 nm),计算得到C―Cl和C―Br S1解离的速率常数分别为3.54 X 1011S-1 和5.82×1011S-1,与实验推测的248 nm时的反应速率~1012S-1稍有不同。
在S1势能面上,BrCH2COC1分子的C―C1 α和C一Br β键解离的反应能垒分别为34.3和23.8kJ/mol(8.2和5.7 kcal/mol,MR-CI/cc-pVDZ//CAS(8.7)/6-31G* )。从能量上看是一对竞争反应,且C―Br β键的解离稍微有利,这与实验结果是不一致的。实验观测表明 ,在248 nm激光激发的条件下,BrCH2CH2COC1分子中,由n→π・跃迁导致的C―Br和C―Cl键解离的支化率小于0.05:1.0,这个数字比BrCH2COC1中的相应值0.4:1.0要小得多。既然C―Cl和C―Br键在S 势能面上的解离只有很小的能垒,那么分子内的能量弛豫在C―Clα和C―Br β键的选择性解离中可能会发挥重要作用。分子内部的能量在传递到C― Br β键之前,C―Cl α键可能已经解离了。这一点很可能是造成较强的C―Cl α键比较弱的C―Br β键更容易解离的原因。
为了证实这一点,以CAS(10,8)/cc-pVDZ计算得到的S1的能量、能量梯度和Hessian矩阵为基础,对S1的Franck-Condon(FC)点做了直接分子动力学计算,如图4所示。
|
|
|
|
|
(a)C―Cl |
(b)C―Br |
(C)C―C |
|
图4 n→π・激发下,BrCH2COC1分子的S.Franck-Condon结构的原始弛豫动力学[54] |
||
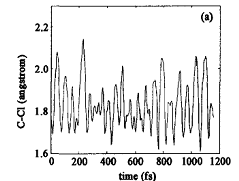
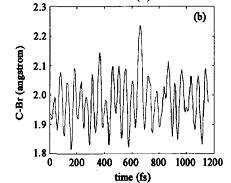
从图4(a)可以看出,轨线从S1FC结构开始,C―Cl键的键长由此时的0.1821 nm很快弛豫到S1极小的0.1786 nm。在初始激发60 fs之后,轨线到达C―Cl S1解离的过渡态区域,此时C和Cl之间的距离为0.205―0.215 nm。事实上,轨线在1 200 fs的传播时间内,多次到达C―Cl键S1态解离的过渡态区域。初始激发是定域在C=0区域,其中部分激发能传递给C―Cl键,导致C―Cl处于高振动激发态,从而使得C―Cl键很快发生解离。图4(b)给出的是C―Br键键长随时间的变化情况。在S1极小和过渡态中,C―Br键的键长分别是0.1950和0.2360 nm,从图4(b)可看出轨线几乎没有机会到达C―Br S1解离的过渡态区域,C―Br键的伸缩振动始终处于基态或者低振动激发态。不同于C―Br键的伸缩振动,初始激发能能够很快传递到C―C键,并使得C―C键的伸缩振动处于高振动激发态,如图4(C)所示。但是由于C―C S1解离的能垒高达145.5 kJ/mol(34.8 kcal/mo1),比C―Cl解离的能垒要高得多,C―C S1解离发生的机会很小。直接分子动力学计算预测,大部分初始激发能传递给口C―Cl伸缩振动,导致C―Cl键在飞秒时间尺度上发生解离;但是初始激发能要通过α-C―C键才能传递到 β-C―Br键,这一过程在几个飞秒时间内完成的几率非常小。因此,在n→π・激发下,分子内能量传递对距离的依赖性是影响BrCH2COC1分子中C―Cl α和C―Brβ键的选择性解离的重要因素。
1.3 甲酸(HCOOH)和乙酸(CH3COOH)
HCOOH是最简单的羧酸,也可以看成是不对称取代的羰基化合物。光诱导的HCOOH的解离和异构过程实验上已经广泛地研究过。我们研究小组结合CASSCF和MR-CI计算对其反应机理从理论上进行了详细的讨论[42],发现光诱导HCOOH的光解离具有波长依赖和构型记忆特性。
在CAS(10,8)/cc-pVTZ水平上优化得到的S0S1FC区域内极小能量交叉点S0/S1、S1/T1结构如图5所示。
|
|
|
图5 HCOOH基态、激发态和交叉点的结构示意图及主要结构参数 (键长:×10 nm;键角:度)[42] |
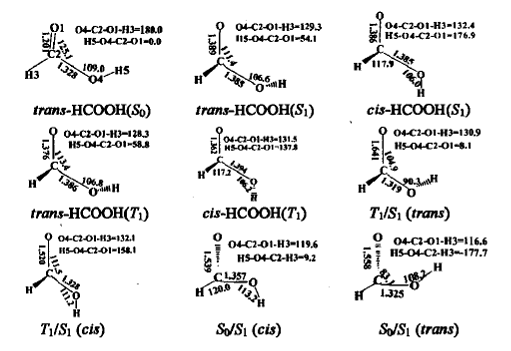
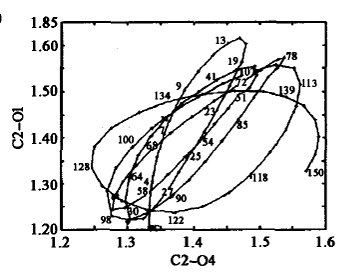
将S1和S0平衡结构进行比较,发现最大的变化是C2一O1、C2一04键长和O1一C2―04键角。S1 FC结构的微分耦合矢量主要对应于C2一O1、C2一04的伸缩振动和04一C2一O1的弯曲振动模式,这将导致体系达到S1平衡结构;而梯度差矢量主要与C2一O1距离的显著降低和O4一C2一O1角度的增大相关,将使HCOOH 回到S0态。与S1平衡结构相比,FC区域内的两个交叉点T1/S1(cis)和/S1 (trans)的结构,尤其是在T1/S1 (trans)的结构中,C2一O1键大幅度伸长,C2一O4键则显著缩短。与S1态FC结构相比,S0/S1结构发生的较大变化主要与C2一O1键长和04一C2一O1一H3二面角相关。在发生垂直激发时,有一个很大的力驱使C2一O1伸长、O4一C2一O1发生弯曲。在FC(S1)、trans-HCOOH(S1)极小和S0/S1 (trans)结构中,C2一O1距离分别是0.1201、0.1389和0.1558nm。直接分子动力学计算得到的分子弛豫过程如图6所示。
|
|
|
|
(a)C―O键的键长随时间的变化; |
(b)二面角随时间的变化[42] |
|
图6 HCOOH S1 FC结构开始的弛豫过程, |
|
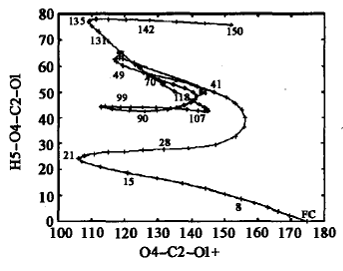
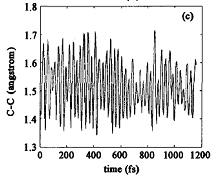
不难发现轨迹在到达S,极小和C2一O4断裂之前已经多次到达S0/S1 (trans)锥形交叉区域。态平均的CAS(10,8)/cc-pVTZ计算确认,S0/S1 (trans)结构是S0与S1势能面的交叉点。经过锥形交叉发生S1→S0内转换过程的时间尺度与振动周期在同一数量级上。在S0/S1 (trans)结构附近,具有高的内转换效率,使得193 nm光激发的trans-HCOOH(S0)很容易回到基态。随后在基态上发生消除反应,形成CO和CO2等产物。
当激发光波长为260-248 nm时,对应的激发能为460-481 kJ/mol(110-115 kCal/mo1)。此时S1态上的直接解离、经过T1/S1交叉点发生系间窜跃和经过S0/S1发生内转换过程在能量上都是不可及的。这种情况下,体系到达S1态以后,主要通过振动相互作用回到基态。n→π・跃迁导致锥形S1平衡构型,其正则振动模式不仅会发生转移和变形,而且相互之间也会发生混合。CAS(10,8)/cc-pVTZ频率计算显示,相对于基态振动模式和频率,S1态甲酸分子的振动频率发生很大的变化,而且C1一O2伸缩振动与H3一C2一O1和O1一C2一O4弯曲振动有很强的混合。振动频率的变化与振动模式的混合增加了内转换过程速率常数中振动部分的贡献,从而使得S1→S0内转换的效率大大提高[56]。一旦弛豫到基态,体系有充足的内能导致CO2+H2 和CO+ H2O的生成。在可能的基态反应途径中,HCOOH→HCO + OH、HCOOH→H + COOH 和HCOOH→HCOO + H三个解离通道是强吸热的,它们无法与上述分子消除相竞争。因此,248 nm光激发HCOOH主要通过振动相互作用内转换到S0并发生分子消除反应。时间分辨FTIR光谱[57]检测到大量振动激发的CO和CO2,这与计算结果是一致的。
当激发光波长为240-210 nm(498-569 kJ/mol,119-136 kCal/mo1)时,在S1势能面上直接解离成HCO( 2A’ )+ OH( 2Ⅱ)以及经由T1/S1交叉点发生系间窜跃是能量上有利的途径,而其中S1→T1的系间窜跃(ISC)过程是自旋禁阻的。根据计算的自旋-轨道耦合矩阵元和T1/S1 (cis )的能量梯度差,利用Landau-Zener定律,计算得到S1一T1ISC过程的几率因子大约是5.5 X 10-4。也就是说,如果将ISC按照自旋允许的过程来处理,S1→T1 的ISC速率将降低104 倍。因此,在S1态上直接发生解离生成HCO(2 A’ )+ OH(2II)是HCOOH(S1)的最可能去活化途径,S1→T1 系间窜跃无法与S1直接解离相竞争。这就解释了为什么在激发光波长为220、222、225和212.8 nm时,实验上观察到OH 自由基高产率地形成[58-68]。
当激发光波长为193 nm(619 kJ/mol,148kCal/mo1)时,经由 T1/S1 发生系间窜跃、经由S0/S1发生内转换或者在S1态上直接解离从能量上来说都是允许的。当激发能为13.0 eV(1 254 kJ/mol,300 kCal/mo1)时,实验上并未观测到COOH和H产物碎片。HCOOH初始光激发主要定域在C=0区域,在内部能量弛豫到 β-O―H键之前,α-C―O键断裂可能已经发生。而且由于能垒较高,C―H S1解离是不能与C―OH S1解离相竞争的。事实上,当激发能为12 ev(1 158 kJ/mol,277 kCal/mo1)时[69],观测到了HCOOH的VUV解离中形成的HCO0自由基的发射谱。因此,在激发光波长为210―193 nm时,HCOOH(S1 )→HCO(2A’ )+ OH( 2II)是能量上最有利的去活化途径。非绝热直接动力学计算表明,HCO + OH解离途径占产物产率的70%[70]。
在248和193 nm激光照射下,trans-HCOOH的分子消除反应途径中CO+H20优先于CO2+H2,而193 nm激光照射下,cis-HCOOH的光解产物中C02+H2是主要产物[71]。很显然,实验结果显示,HCOOH的光诱导分子消除具有结构记忆的特点。这可以通过S0势能面上的驻点结构进行阐释。但是,经典轨迹和RRKM计算[72]对此给出了不同解释,认为S1势能面的形状在决定S1势能面上的短寿命动力学中可能起到重要作用。在193 nm激光照射下,基态HCOOH分子被激发到S1势能面上,得到一个与|s 相对应的结构。在S1 FC结构中,非零能量梯度的方向控制初始激发能在C=O伸缩、C―H面外摇摆和O―H扭转模式中的再分布。仅仅放开C=O伸缩振动进行结构优化,可得到平面的cis-HCOOH(S1)或trans-HCOOH(S1)(二级鞍点)。当去掉对C―H摇摆和O―H转动自由度的限制后,平面trans-HCOOH(S1)主要弛豫到trans-HCOOH(S1)极小。但是,由于存在O―H非零变形力,所以平面trans-HCOOH(S1 )驰豫到cis-HCOOH(S1 )极小的几率也很大。如图6(b)所示,经过150 fs的弛豫后,H5一O4一C2一O1二面角从S1 FC结构中的0°增加到~80°,这与S1的trans-cis异构过渡态中的87.4°非常接近。在S1态上发生trans-cis异构的时间尺度大约在皮秒数量级上。S1的trans-cis异构可能对HCOOH光解过程中的部分构型记忆起到主要作用。
光诱导的乙酸光解机理也得到了从头算水平上的理论研究 [48]。其α-C―O键的S1解离能垒被预测为64.0 kJ/mol(15.3 kCal/mo1),与HCOOH的相应能垒非常接近。在乙酸光解实验中观测到C―O键解离的碎片是各向同性分布的,这可能是由于S1解离的出口能垒比较高造成的。当激发光波长在200-222 nm 范围内时,CH3COOH 主要分布在S1态,此时体系有足够的内部能量克服C―O键解离的能垒而在S1势能面上进行直接解离。C―O S1直接解离是气相乙酸紫外激发条件下形成OH(2II)自由基的一个主要途径。
CH3COOH(S1 )极小和交叉点S1/T1在结构上的相似性说明S1→T1系间窜跃过程在很大程度上是可以发生的。这一点也可以从计算得到的旋轨耦合(SOC)常数获得证据,该耦合常数在S1/T1结构时为68.2 Cm-1。S1/T1交叉点可视为S1→T1非绝热过程的过渡态,其能垒为26.3 kJ/mol(6.3 kCal/mo1),较C―O S1直接解离的能垒低37.6 kJ/mol(9.0kCal/mo1),这表明S1→T1系间窜跃过程与C―O S1直接解离是相互竞争的,ISC到T1并在T1势能面上进行C―O键的解离是获得OH(2 II)自由基的另一个重要途径。即:
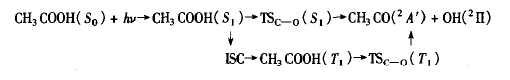
这就解释了气相乙酸光解的主要产物是OH( 2II)和CH3CO( 2A’ )的实验事实。α-C―C键的解离也是在T1势能面上进行的。可见,尽管CH3COOH 和HCOOH是同系物,但二者的光解机理存在较大的差异。
理论研究表明,对于非对称取代的脂肪族羰基化合物,α键的相对强度只是影响α键选择性解离的因素之一,α键的选择性解离主要是由解离机理决定的。如果脂肪族羰基化合物的两个α键解离所产生的基态碎片是非简并的,那么S0和T1态的解离将产生基态碎片,而S1势能面上α解离很可能与激发态碎片绝热相关。在这种情况下,S0、S1和T1 势能面上的解离能垒的相对高度与α键的相对强度大致相当。根据统计过渡态理论,如果不同的键解离途径具有可比的指前因子,那么两个α键中较弱的一个将更容易解离。如果仅有一个α键的解离生成一个双重或者三重简并的基态碎片(Cl或者OH),那么这个α键在S0、S1和T1势能面上的解离将总是生成基态碎片。根据Hammond原理,在S1势能面上与基态碎片绝热相关的α键解离有较低的能垒,这个α键解离优先发生。实际上,在n→π・跃迁的情况下,定性的态相关图可用来判断α键解离的选择性。
非对称取代的脂肪族羰基化合物的光解离可概括为3类:一类是酰卤化合物(RCOX,X=C1、Br、I;R=H、CH3、CH3CH2等),这类化合物的S1态是排斥态或者S1势能面上的a解离仅有非常小的能垒,C―X键在S1态解离非常迅速,系间窜跃以及其它的过程是不能与S1直接解离相竞争的。在这种情况下,α键的相对强度对键的选择性解离没有影响。另一类羰基化合物如CH3CHO、CH3COCN、CH3CONH2、CH3COCH2CH3等,由于能垒较高,这些分子发生S1直接解离的几率比较低。C―C、C―H或C―N α键的解离通常发生在系间窜跃到T1势能面之后。于是,对于这类分子来说,在低能量激发的条件下,两个α键中强度相对较弱的一个优先解离。第三类羰基化合物的代表是CH3COOH,其C一0键在S1态直接解离与系间窜跃到T1势能面相互竞争。这种情况下,两个α键的相对强度只是影响键的选择性解离的其中一个因素,α键解离的选择性主要由光诱导的解离机理和光解动力学决定。
2 芳香族羰基化合物的光解反应
在芳香族羰基化合物中,由于苯环与羰基之间的共轭相互作用,其电子激发态的相对能量和反应性能都显著地不同于相应的脂肪族羰基化合物。实验研究已经发现,处于1nπ・和3nπ・电子态的脂肪酮类化合物都可以发生NorrishⅠ和NorrishⅡ反应,但大多数芳香酮或者芳香醛类化合物(PhCOR)却只能在最低三态上发生上述反应[2-4];气态芳香酮的激发单重态寿命比相应的脂肪酮要短得多;芳香族羰基化合物一般具有强的磷光,但是荧光很弱[73]。这些实验事实说明芳香族羰基化合物的系间窜跃(ISC)过程是非常有效的。研究表明,PhCOR的S1和T1态均起源于羰基的n1π・1电子组态。在这种情况下S1→T1系间窜跃过程电子的自旋和轨道总角动量不守恒,因此PhCOR直接发生S1 (1nπ・ )→T1(3nπ・)系间窜跃的效率应该很低。在凝聚相中存在环境与体系的相互作用,S1 (1nπ・)→T1 (3nπ・)系间窜跃可以高效率地发生。但为什么在气态环境中也能观测到高效的S1 (1nπ・)→T1 (3nπ・)过程?对于芳香族羰基化合物,其T2、T1和S1电子态的能量非常接近,那么T2在系间窜跃过程中是否扮演着重要的角色?针对这些问题,人们对一系列芳香族羰基化合物进行了理论研究,得到了一些非常有意义的结论。
对一系列芳香族羰基化合物(PhCOR,R=H、CH3、CH2CH3、CH2CH2CH3)的几个最低电子态及其极小能量交叉点从理论上进行研究[44]时,发现了S1/T2/T1 三面交叉点。而且研究发现,所研究的芳香族羰基化合物普遍存在S1/T2/T1三面交叉点,并具有几乎恒定的几何结构和电子结构。该交叉点在芳香族羰基化合物的弛豫动力学和光化学反应中应该具有非常重要的作用。由于对芳香族羰基化合物的理论研究是最近才开始的,而且其具有与脂肪族羰基化合物不同的特性,因此我们对这一部分的介绍较为详细。
表1列出了CAS(10,9)/6-31G*计算得到的一系列芳香族羰基化合物的最低5个电子态的绝热激发能。Molina和Merchan" 在CASFT2水平上计算了苯甲醛(C6H5CHO,BA)的S1、T1、S2和T2 的绝热激发能,分别为3.07、3.27、3.16和4.72 eV。虽然CASFT2方法考虑了更多的电子相关,但是计算得到的数据与实验相比并不比CAS(10,9)水平上的来得精确。为了探究烷基链的长度和取代基对电子激发态的结构、能量以及绝热激发能的影响,也对其它一些芳香族羰基化合物进行了计算,结果参见表1。
表1 CAS(10,9)/6-31G*计算得到的一系列芳香族羰基化合物的绝热激发能及相关的实验数据(单位:eV)
|
|
S0 |
T1 |
S1 |
T2 |
S2 |
|
BA |
0.0 |
3.13 |
3.29 |
3.22 |
4.58 |
|
PA |
0.0 |
3.20 |
3.35 |
3.27 |
4.59 |
|
o-CI-PA |
0.0 |
3.02 |
3.18 |
3.28 |
4.53 |
|
m-CI-PA |
0.0 |
3.19 |
3.34 |
3.26 |
4.55 |
|
p-CI-PA |
0.0 |
3.18 |
3.33 |
3.26 |
4.56 |
|
o-CH3-PA |
0.0 |
3.16 |
3.33 |
3.26 |
4.56 |
|
m-CH3-PA |
0.0 |
3.21 |
3.25 |
3.23 |
4.47 |
|
p-CH3-PA |
0.0 |
3.22 |
3.36 |
3.25 |
4.59 |
|
PE |
0.0 |
3.27 |
3.42 |
3.28 |
4.59 |
|
Exp.PA |
0.0 |
3.20 |
3.38 |
(3.30) |
1.39 |
|
(BA)* |
0.0 |
(3.12) |
(3.34) |
(3.30) |
(4.36) |
括号里面的是BA 的实验值。实验值来自文献73、74。
结果表明,不同取代基以及取代位置对绝热激发能的影响不大,这预示着更多的芳香族羰基化合物其基态到几个低电子激发态的绝热激发能近乎是常数。通过与实验得到的0-0能量进行比较可知,CAS(10,9)/6-31G* 计算能够非常好地重现芳香族羰基化合物的绝热激发能。
芳香族羰基化合物中含有相对较大的共轭体系,致使其π轨道和σ轨道能量相差较大,从而能量分离情况比较好。在CASSCF计算中,将近简并的π轨道和氧原子的非键轨道全部包含在活化空间中,即可以比较精确地预测芳香族羰基化合物电子激发态的结构和相对能量。尽管MR-CI和CASPT2方法在考虑动态电子相关方面更为有效,但是不经优化而只是进行MR-CI和CASPT2单点能量计算,会带入一些误差而降低整体的精度,因为电子激发态对电子相关比基态更为敏感。CASSCF和CASPT2方法可用于优化小的有机分子,但是CASPT2优化得到的结构参数与实验相比并不会比CASSCF的结果有显著改善。而CASPT2方法优化几何结构是非常耗时的,而且目前为止尚不能用于较大的体系。事实上,只要活化空间选择合理,CASSCF会是计算中等大小分子的激发态势能面的最好选择。
计算表明,所研究的芳香族羰基化合物的激发态几何和电子结构具有一定的共性。其S1、T1、S2和T2态分别具有1nπ・、3nπ・、1ππ・和3ππ・性质。基态的芳香族羰基化合物中,苯环几乎是一个正六边形。与基态相比,S1和T1在结构上有很大变化,主要表现为C―O键的伸长,其键长由基态时的~ 0.121 nm增加到~0.135 nm。n→π・电子跃迁主要定域在C=O部分,对苯环的结构影响很小。众所周知,n→π・电子跃迁导致脂肪族羰基化合物形成锥形的S1和T1极小结构,此时羰基碳原子采用sp3杂化。而在芳香族羰基化合物中,由于苯环与C=O的共轭作用,其S1和T1结构是平面的。芳香族羰基化合物的S2和T2对应于n→π・电子跃迁,主要定域在苯环上,对苯环部分的结构有较大影响,而对C=O部分几乎没有影响。S2极小中两个未成对电子离域到整个苯环中,而T2极小中的两个未成对电子则主要定域在与羰基相连的C及其对位C原子上,这与实验结果[75]是一致的。
如前所述,在芳香族羰基化合物的S1 或T1 结构中,两个未成对电子主要分布在羰基的C和0原子上,在T2中则主要分布在苯环上。计算表明,交叉点S1/T2 和 T2/T1 ,在结构和能量上都是无法分辨的,表明芳香族羰基化合物(PhCOR,R=H、CH3、CH2CH3、CH2CH2CH3)的S1、T1 和 T2势能面在同一个区域交叉。如图7所示,
|
|
|
图7 芳香族羰基化合物S1(T1)、 T2和S1/T2/ T1的结构示意图 |
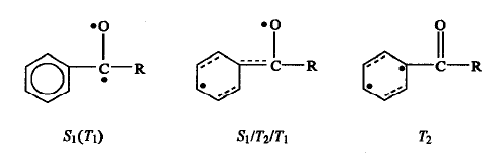
S1/T2/T1,结构介于S1(T1)和 T2之间,两个单占电子分别分布在0原子和苯环上。S1/T2/T1的结构差别在于共轭π电子进行重新分布(参见表2)。
表2 PhCOCH3的S1 / T2 (1nπ・/3ππ・)和T1 / T2 (3nπ・/3ππ・ )
交叉结构的能量和单电子密度(密度矩阵的对角元)[44]
|
|
S1/T2 |
T1/T2 |
||
|
1nπ・ |
3ππ・ |
3nπ・ |
3ππ・ |
|
|
π1 |
1.93 |
1.86 |
1.93 |
1.86 |
|
π2 |
2.00 |
2.00 |
2.00 |
2.00 |
|
π3 |
1.94 |
1.04 |
1.94 |
1.04 |
|
π4 |
1.94 |
1.91 |
1.94 |
1.91 |
|
π |
1.00 |
2.00 |
1.00 |
2.00 |
|
π5・ |
0.99 |
0.95 |
0.99 |
0.95 |
|
π6・ |
0.07 |
0.09 |
0.07 |
0.09 |
|
π7・ |
0.13 |
0.16 |
0.13 |
0.16 |
|
E(a.u.) |
-328.4206720 |
-328.4206719 |
||
目前为止研究的所有芳香族羰基化合物的PhCO部分的结构都是类似的,S1/T2/T1三面交叉点在芳香族羰基化合物中极可能是普遍存在的,而且具有几乎恒定的几何结构和电子结构。类似的规律在多烯和多烯自由基的光异构化反应[76]中也发现过,在反应中起重要作用的锥形交叉点S1/S0也具有相同的几何构型和电子特征。
PhCOR 的T2态主要具有3ππ・性质。在S1 (1ππ・)→T2 (3ππ・)系间窜跃过程中,自旋角动量的改变伴随着轨道角动量的改变,而后者有利于产生强的自旋一轨道耦合,从而使得这一过程具有很高的效率。对于苯乙酮和苯甲酮,实验测得其S1→T2过程的速率为~1011s-1。T2和T1态之间可以通过交叉点T2/T1进行内转换,这一过程可以在转动周期内完成。S1 /T2/T1的存在导致S1→T1过程可以借助T2态而顺利地完成。尽管直接的S1 (1nπ・)→/T1(3nπ・)系间窜跃由于不能发生一级自旋-轨道耦合而效率极低,但是借助T2态的传递,S1 (1nπ・)→/T1(3nπ・)系间窜跃则可以高效率地完成。这就解释了芳香族羰基化合物的S1寿命较脂肪族羰基化合物短得多,光化学反应总是起源于三态,而且磷光强、荧光弱的实验事实。另外,实验已经发现,大量的芳香酮类化合物发生系间窜越的速率常数(kst)大约为1011s -1[6,77-81],而脂肪酮类化合物的kst在107―108 s-1 范围内[82-83]。S1 /T2/T1交叉点的存在有助于解释芳香酮类化合物和脂肪酮类化合物的kst之间为什么会有1 000倍的差别。
3 光化学反应从头算研究展望
光化学反应机理的从头算研究在近几十年中得到了快速的发展。如前所述,光化学反应过程涉及到电子激发态,建立在单参考态基础上的从头算方法一般很难正确地描述电子激发态的特性。多参考态的电子相关方法目前只能用来研究非常小的分子体系。CASSCF方法能够平衡地描述基态和激发态的结构,目前广泛地被用来优化激发态结构和反应途径。但是CASSCF方法对活化空间的选择有相当的要求,活化空间中包含的活化轨道不同会对计算结果特别是能量产生很大影响。这使得CASSCF计算对经验、技巧和策略甚至目标体系都有很大的依赖性。CASSCF方法仅包含了部分电子相关效应,在能量的计算上与实验结果尚有一些误差。一个补救的措施是在CASSCF优化结构的基础上,用MR-CI和CASPT2等高精度的电子相关方法进行单点能量计算。CASSCF和MR-CI组合方法是目前光化学反应机理从头算研究一种合适的选择。光化学反应机理从头算研究的一个重要方向是发展快速和精确的电子相关方法。另外,目前光化学反应的从头算研究主要集中在气相,而现实中很多光化学反应是在溶液中或者固相进行的。从理论上精确地考虑溶剂或者固相环境对光化学反应过程的影响,是另一个重要的发展方向。另外,光化学反应在生命过程、环境保护、新材料、新能源和信息技术等领域中发挥着独特的作用。对光生物过程和光电功能材料性能的计算模拟也是一个活跃的研究领域。目前,已经有研究小组采用不同的策略对生命过程中涉及的光化学反应过程进行了研究,并取得了一些结果[84-90]。
参考文献
[1] De Fea r S,Diau E W C,Zewail A H.Angew.Chem.Int.Ed.,2000,39;26 -263
[2] Wagner P J.Acc.Chem.Res.,1971,4;168-177
[3] Gilbert A,Baggott j.Essentia1s of Mo1ecu1ar PhotoChemistry.Boca Raton,FL;CRC Press,1991
[4]Horspoo1 W, Armesto D. Organic Photochemistry; A Comprehensive Treatment.Ellis Horwood limited,1992
[5] Wagner P J.in CRC Handbook of Organic Photochemistry and Photobio1ogy(eds.Horspoo1 W M,Song P S).Boca Raton;CRC Press,1995
[6] Turro N J.Modem Mo1ecular Photochemistry.University Science Books, 1990
[7] Diau E W C,Kftting C,Zewail A H.Chem.Phys.Chem,2001,2;273-293
[8] Diau E W C,Kotting C,Zewail A H.ChemPhysChem,2001,2;294-309
[9] Liu D,Fang W H,Fu X Y.Chem.Phys.Lett.,2000,325;86-92
[10] Setokuchi O,Simizu Y.J.Mol.stract.,1997,401;29-33
[11] Setokuchi O,Matuzawa S,Shimizu Y.Chem.Phys.Lett.,1998.284;19-23
[12] Sakurai H,Kato S.j.Mo1.stmCt.,1999,461;145-152
[13] Diau E W C,Kstting C,Se11ing T I,et al.Chem.Phys.Chem,2002.3;57-78
[14] Hess B,Bmna P J,Buenker R J,et al.Chem.Phys.,1976,18;267-28O
[15] Ga1asso V.J.Chem.Phys.,1990,92;2495-2504
[16] Gwa1tney S R,Bart1ett R J.Chem.Phys.Lett.,1995,241;26-32
[17] Merchan,Roes B O,MC Diardm1d R,et al.J.Chem.Phys.,1996.104;1791-1804
[18] Liao D W,Mebel A M,Hayashi M,et al.J.Chem.P1ays.,1999,111;205-215
[19] Solling T I,Diau E W C,Kott1ng C,et al.ChemPhysChem,2002 3;79-97
[20] Deshmukh S,Hess W P.J.Chem.Phys.,1994,100;6429-6433
[21] Deshmukh S,Myers J D,Xantheas S S,et al.J.Phys.Chem.,1994.98;12535-12544
[22] Sumathi R,Chandra A K.J.Chem.Phys.,1993,99;6531-6536
[23] Arunan E.J.Phys.Chem.A,1997,101;4838- 4839
[24] Lane I C,Meehan R,Powis I.J.Phys.Chem., 1995,99;12371-12374
[25] Kroger P M,Ri1ey S J.J.Chem.Phys.,1977,67;4 83- 490
[26] PersonM D,Kash PW,Bufer 1 J.J.Chem.Phys.,1992,97;355― 373
[27] PersonM D,Kash PW,Butler L J.J.Phys.Chem.,1992,96;2021-2023
[28] North S,Blank D A,Lee Y T.Chem.Phys.Lett.,1994,224;38-42
[29] Hunnicutt S S,Waits LD,Guest J A.J.Phys.Chem.,1989,93;5188-5195
[30] Hunnicutt S S,W aits L D Guest J A.J.Phys.Chem.,1991 95;562-570
[31] Naik PD,Upadhyaya H P Kumar A,et a1.Chem .Phys.1ett 2001.340; 116-122
[32] PersonM D,Kash PW ,SChofie1s S A,et a1.J.Chen1.Phys.,1991,95;3843-3846
[33] Kash P W ,Waschewsky G C G,But1er 1 J.J.Chem.Phys.,1994.10o;4017- 4018
[34] Kash PW ,WasChewsky G C G,Bufer 1 J.J.Chem.Phys.,1993.99;4479-4494
[35] WaschewskyG C G,Kash P W ,Myers T L,et al.J.Chem.Soc.Fraraday Trans.,1994,90;1581-1598
[36] But1er L J.Annu.Rev.Phys.Chem.,1998,49;125-171
[37] Marks A J.J.Chem.Phys.,2001,114;1700-1708
[38] Chen X B,Fang W H.J. Am.Chem.Soc.,2004, 126;8976-898o
[39] Chen X B, Fang W H.J.Am.Chem.Soc., 2003, 125;9689-9698
[4o] Chen X B,FangW H,Phi11ips D L.J.Phys.Chem.A,2006,1 10;4434-4441
[41] Wang Y W,He H Y,Fan g W H.J.Mo1.StruCt.,20o3,634;281-287
[42] He H Y,Fang W H.J.Am.Chem.Soc.,2003,125;16139-16147
[43] He H Y,Fang W H.J.Phys.Chem.A,2004,108;5386-5392
[44] Fang W H ,Phillips D L.ChemPhysChem,2002,10;889-892
[45] Fang W H,Phi11ips D 1.J.Theor.Comput.Chem.,2003,2;23-31
[46] Fang W H, Liu R Z.J.Chem.Phys.,2001,115;10431-10437
[47] Tian Y C,Fang W H.J.Phys.Chem.A,2006,110;11704-11710
[48]Fang W H,Liu R Z,Zheng X M,et al.J.Org.Chem.,2002,67;8407-8415.and references therein
[49] Xiao H Y,Liu Y J,Fang W H.J.Mo1.Struct.,2007,802;99-103
[50] K J,Zhang F,Fang W H.J.Phys.Chem.A,2005,109;7718-7724
[51] Ding W J,Fang W H,KuR Z.Chem.Phys.1ett.,2003,369;57 -578
[52] Ding W J,Fang W H,KuR Z.Chem.Phys.1ett.,2002,351;9-17
[53] Ding W J,Fang W H,ku R Z,et al.J.Chem.Phys.,2002,117;8745- 8753
[54] Zhang F,Ding W J,Fang W H.J.Chem.Phys.,2006,125;art.no.184305
[55] Chen S L,Fang W H.J.Phys.Chem.A,2006,110;944-950
[56] Mebel A M,Hayashi M,Liang K K,et al.J.Phys.Chem.A,1999, 103;10674-10690,and referenCes therein
[57] Su H M,He Y,Kong F A,et al.J.Chem.Phys.,2000,113;1891-1897
[58] Jo11y G S,McKenney D J,Singleton D 1,et al.J.Phys.Chem .,1986,90;6557-6562
[59] Singleton D L,Paraskevopou1os G,Irwin R S,et al.J.Am.Chem .Soc.,1988,110;7786-7790
[60] Singleton D L,Paraskevopou1os G,Irwin R S.J.Am.Chem.SoC.,1989,111;5248-5251
[61] Jolly G S,Singleton D L,McKenney D J,et al.J.Chem.Phys.,1986,84;6662- 6667
[62] Jolly G S,Singleton D 1,Paraskevopou1os G.J.Phys.Chem.,1987。91;3463― 3465
[63] Singleton DL,Paraskevopoulos G,Irwin R S.J.Phys.Chem.,1990.94;695-699
[64] Lrwin R S,Sing1eton D L,Paraskevopoulos G,et al.Int.J.Chem.Kinet.,1994 ,26;219-225
[65] Ebata T,Amano T,ho M.J.Chem.Phys.,1989,90;112-117
[66] Brouard M.O,Mahony J.Chem.Phys.Lett.,1988,149;45-50
[67] Brouard M,Simons J P,Wang J X.Faraday Discuss.Chem.Soc.,1991,91;63-72
[68] Lee K W,Lee K S,Jung K H,et al.J.Chem.Phys.,2002,117;9266-9274
[69] Schwe1 M,Dulieu F,Jochims H W,et al.J.Phys.Chem.A,2002.106;10908-10918
[70] Martinez-Nfifiez E,Vfzquez S,Granucei G,et al.Chem,Phys.1ett.,2005,412;35-40
[71] Khriachtchev L,Macoas E,Pettersson M,et al.J.Am.Chem.Soc.,2002,124; 10994-10995
[72] Garavelli M,Bernardi F,Olivucci M,et al.Faraday.Discuss.,1998.110;51-70
[73] Silva C R,Reilly J P.J.Phys.Chem.,1996,100;17111-17123,and References Therein
[74] Molina V,Merehan M.J.Phys.Chem.A,2001,105;3745-3751.an d References Therein
[75] Feenstra J S,Park S T,Zewai1 A H.J.Chem.Phys.,2005,123;221104
[76] Bemardi F,Olivucci M,Robb M A.Chem.Soc.Rev.,1996,321-328.and References Therein
[77] Anderson R W,Hochstrasser R M,Lutz H,et al.J.Chem.Phys.,1974,61;2500-2506
[78] Morris J M,Williams D F.Chem.Phys.1ett.,1974,25;312-314
[79] Matsumoto T,Sato M,Hirayama S.Chem.Phys.1ett.,1972,13;13-15
[80] El-Sayed M A.J.Chem.Phys.,1964,41;2462-2467
[81] El-Sayed M A,Leyerle R.J.Chem.Phys.,1975,62;1579-1580
[82] Halpern A M,Ware W R.J.Chem.Phys.,1970,53;1969-1977
[83] Hansen D A,Lee E K C.J.Chem.Phys.,1975,62 ;183-189
[84] Koseki S,Sehmidt M W,Gordon M S.J.Phys.Chem.,1992,96;10768-10772
[85] Lumento F,Zanirato V,Fusi S,et al.Angew.Chem.Int.Ed.,2007.46;414-420
[86] Andruniow T,Fantacci S,de Angelis F,et al.Angew.Chem.Int.Ed.,2005,44;6077-6081
[87] Sinicropi A,Andruniow T,Ferre N,et al.J.Am.Chem.Soc.,2005.127; 11534-11535
[88] O1ivucci M,Lami A,Santoro F.Angew.Chem.Int.Ed.,2005.4 ;5118-5121
[89] Groenhof G,Bouxin-Cademartory M,Hess B,et al.J.Am.Chem .Soc.,2004,126;42 28-4233
[90] 李娟(Li J).北京师范大学博士学位论文(Ph.D.Dissertation of Beijing Norma1 University),2007