��������ҩ�����������뼼��yd7009
������Դ�����ǵ���ͼ��ݲ���\<ҩ��ѧ>���İ�\�ϵ�������
�ڰ����Ƽ��¼���(P122-139)\½�����
�Ͱ����ǽ�30����Ӧ����ҩ����¹��ա��¼��������Ʊ�����ͨ���Ͱ�����(microencapsulation)������һ���ϵ������Ȼ�Ļ�ϳɵĸ߷��Ӳ���(ͳ��Ϊ�Ҳ�)��Ϊ��Ĥ�ڿ�(membrane wall)������̬ҩ���Һ̬ҩ��(ͳ��Ϊ������)��������ҩ�����ͽ��ң������(microcapsule)��Ҳ��ʹҩ���ܽ��(��)��ɢ�ڸ߷��Ӳ��ϻ����У��γɻ�����(matrix type)С��״ʵ��Ĺ���Ǽ������(microsphere)���Һ�������������������������ķֱ�������Һ�����������Ҫ�����ҡ�
(һ)ҩ���һ����ص�
ҩѧ��Ѹ�ͷ�չ��ҩ���������深�ࡣ����ԭ���Ƽ������������������ٴ�Ҫ����֮�������¹��ա��¼���Ӧ�����Ƽ��Ʊ������С��һ����ǽ���ҩ��Ӧ���¹��ա��¼����е�һ��ͻ������������30����ҩ���ڽ��������һ��������������ʹҩ�������ء����ġ�����ҩ��ά���ء�����ҩ�Լ������ҩ�ȡ����е��һ���Ʒ�к�ù��Ƭ(����)�������ܲ���Ƭ(��ʿ)�ȡ�ҩ���һ����������ص㣺
(1)�ڸ�ҩ��IJ�����ζ����ζ��������͡��ȱ���������������Լ��ǰ���ȣ�
(2)���ҩ����ȶ��ԣ����������������ܲ��ء���ˮ�����еİ�˾ƥ�֡��ӷ��Ļӷ����ࡢ�����ԡ�ˮ������������Ի�����ҩ�
(3)��ֹҩ����θ��ʧ�����ٶ�θ�Ĵ̼��ԣ�����ø����ù�ء��ȵ��ص�����θ��ʧ��Ȼ��ء����������ȴ̼�θ������θ�����һ��ɿ˷���Щȱ�㣻
(4)ʹҺ̬ҩ���̬������Ӧ�������棺�����ࡢ���ϡ�Һ����֬����ά���صȣ�
(5)���ٸ���ҩ�������仯���簢˾ƥ�����˶��������ɼ��ٰ�˾ƥ�ֵ�ˮ�⣬�ֱ���Һ���Ը��ƣ�
(6)���ͻ����ҩ��ɲ��ö������ʡ���Ĥ(���ԵĻ�����pH�ı�Ĥ)�����オ�����(���������Բ�ͬ�IJ���)����ˮ�������ȣ��Ƴ��ҿ�ʹҩ����ͻ��ͣ����Ƴɿ��ͻ����Ƽ�.
(7)ʹҩ��Ũ���ڰ�����������ָ���͵�ҩ���ϸ����ҩ��(����ҩ)�Ƴ��ҵİ����Ƽ����ɽ�ҩ��Ũ���ڸλ�εȰ����������Ч�����Ͷ������á�
(8)�ɽ���ϸ��������������ʰ��ң����ȵ���Ѫ�쵰�ȣ������ڿɷ�������������ã��Ҿ������õ����������Ժ��ȶ��ԡ�
ֵ�÷�����ǣ���ȥ�����˾����������ɸѡ��ҩ���ɰ���ǧ����ǰ;��ҩ����ѡ������Ϊ�ڷ��Ļ��Ե͡���ע��İ�˥�ڶ̡���������һ���һ�¼�����ҩ���һ���ͨ����θ������ҩ�������ã����ఴ��ȥ����Ϊ���ϸ����ѡҩ������Ƴ��������ҩ�������ҩ�Ŀ��������ر������塣
(��)ҩ���һ������Ľ�չ
Ŀǰ�������һ���ҩ���Ƽ���Ʒ�����࣬��ҩ���һ��������о�ȴ��ͻ���ͽ���
Ӣ�������Journal of Microencapsulation��־(����)��l 984�괴����������1999���ѳ���16����רҵ���һ����ʻ���ÿ2���ٿ�һ�Σ�1999��9�½���Ӣ�����ٿ���ʮ�����һ����ʻ��飮�����ࡢ������ҩ����һ����������オ����ϵ�Ӧ�ã�����ҩ����ͷţ��о��ҽṹ���·��������ܼ����¹��գ���̬�������Ĵ��ģ�����Լ������ҡ����������ڿڷ����β����θ����ճĤ�ȷ��棬����������չ������һ��о���Ӧ�ö��ܴ�Ĵٽ����á�
�һ������Ľ�չ�ɷ�Ϊ������[22]�� 80�����ǰ��ҪӦ������Ϊ5µm-2mm���ҡ�80�����չ������С(0.0l-10µm)�ĵڶ�����Ʒ�������Ʒͨ����θ������ҩʱ�������ٻ���֯�����������ӳ�ҩЧ�����Ͷ��ԡ�����Ժ��������ö�.��������Ʒ��Ҫ�������������ӵİ����Ƽ��������г�������պ����ò�λ���Ƽ�������ʮ���¡�
������ţ�オ������ۺ��↑����һЩ����;��ͨ���˹���ѧ˨����ע����Һ��������ܰ�ϵͳ��������������Ƽ����ѳɹ���Ӧ��Ӱϸ��(ghost Cell)������ϸ��(���ϸ��)�����壬ʹ���������Ե��Ը��ƣ����Ϳ��ͱ���ҩ����߿���ζ�(titer)�Ŀ�ԭ�һ�Ҳ��ʵ�֡��ٴ��ѽ��һ�����Ӧ�������е�������ӣ��絰���ʡ�ø�����ء����࣬����Ӧ���ڻ�ϸ�����������������ʧ����ԡ��˹�ϸ��Ӧ��ģ��ϸ������Ȼ���ܣ��ȱ���ԭ�гɷ֣��ֿɽ�����ͷ��������Ľ�����Ӧ�������ҪĤ�ϱ������а��ԣ�Ҳ��Ҫ��Ĥ�ܷ�ֹ���߷�Ӧ����������ʧ����ų�ķ�Ӧ�������Ϳ���Ӧ��ø�����������������ݵĻ�ϸ�������û��Ʒ���Ϊ������֯��ϸ�����ܲ�ȫ�����µ�;�������Ƴ��һ����ȵ������ܱ��ֻ��������������Ķ������ڳ�ʱ�ڲ��Ϸ����ȵ��ء��ٴ��Ϻ���Ӧ�ð��ҵĻ���̿������ѭ��������˥��ι���ʧ���IJ��˽ⶾ���˹���ϸ�����ⷽ���Ӧ��Ҳ����ǰ;���ܲ�������¡������ӽ�ϸ�����ѳɹ��ذ��Ҷ�Ӧ�������﹤�̴����������塣
(��)���������Ҳ�
1. ������ �ҵ�������(Core material)����ҩ�����
��������һ����������븽�Ӽ������ȶ�����ϡ�ͼ��Լ������ͷ����ʵ����ͼ����ٽ�����������Ĥ�����Ե����ܼ��ȡ�����������ǹ��壬Ҳ������Һ�塣ͨ������ҩ�븽�Ӽ����Ⱥ��һ�������Ƚ���ҩ�����һ����ټ��븽�Ӽ������ж�����ҩ���ɽ����������������ɷֱ��һ����ٻ�ϣ���ȡ�������Ҫ��ҩ��Ҳĺ��Ӽ������ʼ����������ȡ����ò�ͬ�Ĺ�������ʱ����������Ҳ�в�ͬ��Ҫ��������������۷�ʱ�����������ܵĻ����ܵľ��ɣ����������۷���Ҫ�������������ˮ���Եġ�����Ҫע�����������Ҳĵı����ʵ�������������٣���������������Ŀ��ҡ�
2���Ҳ� ���ڰ�������IJ��ϳ�Ϊ�Ҳ�(Coating material)�����Ҳĵġ���Ҫ���ǣ�
�������ȶ����������˵���ҩ���ʣ��������̼��ԣ�������ҩ�����顢��Ӱ��ҩ���ҩ�����ü������ⶨ�����С�����ǿ�ȼ������ԣ�����ȫ������������з���Ҫ���ճ�ȡ����ԡ���ˮ�ԡ��ܽ��Եȴ��ԡ����õ��ҲĿɷ�Ϊ���������ࡣ
(1)��Ȼ�߷����Ҳģ���Ȼ�߷��Ӳ�������õ��Ҳģ������ȶ���������Ĥ�Ժá�
1)�����������ǰ��������Ľ����γɵ�ֱ���ۺ��ͨ����Mmw(ƽ��������)��15000-25000֮�䲻ͬ�������Ļ������Ʊ�ʱˮ�ⷽ���IJ�ͬ���������ᷨ����(A��)�ͼ����(B��)��A�������ĵȵ��Ϊ7-9��10g��L��Һ25���pHֵΪ3.8-6.0�� B�������ȶ������׳������ȵ��Ϊ4.7-5.0��10g/L��Һ25���pHֵΪ5-7.4��
���ߵij����������Բ����Һ��ճ�Ⱦ���0.2-0.75cPa��s֮�䣬�����オ�⣬������ԭ�ԣ�ͨ���ɸ���ҩ�������Ե�Ҫ��ѡ��A�ͻ�B�ͣ������Ʊ��ҵ�����Ϊ20-100g/L��
2)����������ϵ�������ἰ��������ļء��ơ�þ������ɡ�һ�㳣�������������ʹ�ã����Ҳĵ�����Ϊ20-100g��L������������������ϲ��ϡ�
3)�������Σ�ϵ������������ϡ��Ӻ�������ȡ���á��������ƿ����ڲ�ͬ�¶ȵ�ˮ�У��������Ҵ������Ѽ������л��ܼ�����ͬMmw��Ʒ��ճ���в��졣������ػ����������������ϲ��ϡ�������Ʋ�����ˮ���ʺ������ƿ���CaCl2�̻����ҡ��о�������������Ժ������ε�Ӱ�죬�������(120�桢20����)ʹ��10g��L��Һ��ճ�Ƚ���64�������¼���(80�桢30����)����ѭ��ʱ���Ч���������ʹ���������ϼ����û����������Ҳ����ճ�ȺͶϼ���Ĥ�˹������IJ���ճ�Ⱥ�Mmw�����䡣
4)�Ǿ��ǣ��Ǿ������ɼ��������������Ƶõ�һ����Ȼ�������Ӷ��ǡ��������������ˮ��Һ��������ԭ�ԣ��������ܱ��ܾ�ø��ø�⣬�������������オ���Ժͳ�Ĥ�ԣ������ڿ����ͳ�ˮ������
(2)��ϳɸ߷��Ӿ۲ģ����Ҳĵİ�ϳɸ߷��Ӳ��϶�ϵ��ά����������ص��Ƕ���С��ճ�ȴ��κ��ܽ������
1)�ȼ���ά���Σ�������ά�������������͵ĸ߷��ӵ���ʣ����ȼ���ά����(CMC-Na)������������������Ҳģ�һ��ֱ���1-5g/L��CMC-Na��30g/L�������ٰ������2��1��ϡ�CMC-Na��ˮ���ͣ����������l 0����������Һ�в��ܡ�ˮ��Һճ�ȴ��п���������һ�������ȶ��ԣ����ᷢ�ͣ�Ҳ�����Ƴ�����CMC-A1�������Ҳġ�
2)������ά��̪������������ά��̪����(CAP)��ǿ���в��ܽ⣬������PH��6��ˮ��Һ�������к������Ȼ�������Ժ���������ˮ��Һ��pHֵ�����ܽ�CAP����Һ�����pHֵ�������Ҳ�ʱ�ɵ���ʹ�ã�����һ����30g/L���ң�Ҳ�����������ʹ�á�
3)�һ���ά�أ��һ���ά��(EC)�Ļ�ѧ�ȶ��Ըߣ������ڶ���ҩ����һ���������ˮ�����ͺͱ��������������Ҵ�����ǿ����ˮ�⣮�ʶ�ǿ����ҩ�ﲻ����.
4)����ά�أ�����ά��(MC)�������Ҳĵ�����Ϊ10-30g��L�������������CMC-Na����άͪ(PVP)������������Ҳġ�
5)�DZ�����ά�أ��DZ�����ά��(HPMC)��������ˮ��Ϊճ����Һ�����������ȶ����б�����ԣ���������(42-56)��105N��cm��
(3)�ϳɸ߷����Ҳģ����Ҳ��õĺϳɸ߷��Ӳ��ϣ��з����オ��ĺ����オ������ࡣ�����オ�⣮�Ҳ���pHֵӰ����Ҳ��о����������ȡ����ﲻ���⣬������һ��pH�������ܽ���Ҳ��о۱�ϩ����֬������ϩ���ȡ������������オ��IJ��ϵõ��㷺��Ӧ�ã����̼�����۰����ᡢ������(PIA)���������ҽ��������������-���Ҷ���Ƕ�ι�����(PLA-PEG)����-��������������������ȣ����ص���������Ĥ�Ժá���ѧ�ȶ��Ըߣ�������ע�䡣
�������������о���ࡢӦ���������オ��ϳɸ߷��ӣ����ǻ����϶����ǻ�����������ľۺ�����õ��ǻ���������(1actic acid)���ǻ�����(g1yco1ic acid)���������ϵõ��ľ�����PLA��ʾ�����ǻ��������ϵõ��ľ�����PGA��ʾ�����������ǻ�����ֱ�����ϵ���PLAGA��ʾ��
�������Գ����ȷ������ⶨ����Ҫ�����Dz������¶�Tg�;����۵�Tc(���ۺ�����һ���̶Ƚᾧ��ʱ)���ȷ������˽���ҩ�ҽṹ����仯��
��Щ�ۺ��ﶼ���ֳ�һ���Ľ��⡢��ʴ�����ԡ������Ǿۺ���ϼ�����������С��ֱ����Ϊ���壻��ʴ��ָ�ֽ��С���������˾ۺ���ᾧ�ȵ͵Ľ���Ͽ졣�������Mmw��Χ��1��-40��������Ϊ2-1 2���£�Mmw��90000���۵�Ϊ60�棬������6���½��⡣�����þ�3-�ǻ�������(PHB)Ϊ�Ҳ��Ƴ��ȵ�����ע�����������3���½��⡣���������������ҽ����������ڹ���ʱ�и��ֱ����������������ҽ�����75��25�Ĺ�����Ϊ�Ҳġ�������1���¿ɽ��⣬��85��15��Ϊ�Ҳģ�������3���½��⡣PLA-PEGǶ�ι�������PLAΪ��ˮ�ԣ�PEGΪ��ˮ�ԣ����ڶ��ߵı������������Mr�ɿ����併�����ܣ����⽵�����ڿɿ�����1-9���·�Χ�ڣ��Ӷ���������ҩ����ͷ����ʡ�
Ӧ�ø߷��Ӹ��Ӽ������������һ��������ɿ����ҵ�ճ���;ۼ�.Ϊ�˿���ע�����ҵ��������ﵽ�й����������Ե�Ҫ��.�Ѷ��˹��ϳɵġ���Ϊ��ˮ�Եĸ߷��Ӳ������˸�Ϊ������о���
�����һ�����
����ҩ����Ҳĵ����ʺ��ҵ��������ͷ������Լ�������Ҫ��ѡ��ͬ���һ��������ɹ���Ϊ������ѧ����������е���ͻ�ѧ�������ࡣ
(һ)������ѧ��
�����һ���Һ���н��У����������Ҳ���һ���������γ��������������ֳ�����뷨(phase separation)�����һ��������ɷ�Ϊ������ķ�ɢ���Ҳĵļ��롢�Ҳĵij������ҲĵĹ̻�4����
�����γ�������IJ�ͬ������뷨�ַ�Ϊ�����۷��������۷����ܼ�-���ܼ������ı��¶ȷ���Һ�и��������빤�����ѳ�Ϊҩ���һ�����Ҫ����֮һ���������豸���߷��Ӳ�����Դ�㷺���ɽ���������ҩ���һ���
1.�����۷� �����۷�(simple coacervation)������뷨�нϳ��õ�һ�֣������ڸ߷����Ҳ�(������)��Һ�м������ۼ��Խ��߷��Ӳ��ϵ��ܽ�ȶ����۳��ҵķ�����
(1)����ԭ�����罫ҩ���ɢ������������Һ�У�Ȼ��������ۼ�(������ǿ��ˮ�Ե���������ƻ�����淋�ˮ��Һ����ǿ��ˮ�Եķǵ�������Ҵ����ͪ)��������������ˮ��Ĥ��ˮ���������ۼ���ϣ�ʹ�������ܽ�Ƚ��ͣ����Ӽ��γ������������Һ�������������γ������ҡ����������ǿ���ģ�һ������ٽ����۵�����(���ˮϡ��)���Ϳɷ��������ۣ�ʹ�����Һܿ���ʧ�����ֿ��������Ʊ������пɼ������ã�������������������ۣ�ֱ���������γ�������״Ϊֹ(���������۲�)������ٲ�ȡ��ʩ���Խ����̻���ʹ֮��Ϊ�����ᡢ��ճ����������������ҡ�
(2)�������̣�����������Ϊ�ҲĵĹ����������£�
|
|
����(��Һ��)ҩ�� |
|
3%-5%������Һ |
|
||||||
|
|
�K�L |
|
||||||||
|
|
�� |
|
||||||||
|
|
����Һ(����״Һ) |
|
||||||||
|
50�� |
�� |
10��������Һ����pH3.5-3.8����60��Na2SO4��Һ |
||||||||
|
|
������ |
|
||||||||
|
|
�� |
��ϡ��Һ* |
||||||||
|
|
������ |
|
||||||||
|
15������ |
�� |
37����ȩ��Һ(��20%NaOH����pH8-9) |
||||||||
|
|
�̻��� |
|
||||||||
|
|
�� |
ˮϴ����ȩ |
||||||||
|
|
�� |
|
||||||||
|
|
�� |
|
||||||||
|
|
�Ƽ� |
|
||||||||
*ϡ��Һ�䷨��ϡ��Һ��Na 2SO4��Һ����Ũ����������ϵͳ�е�Na 2SO4Ũ��(��Ϊa��)��1.5%[��(a+1.5)��]��ϡ��Һ���Ϊ������ϵͳ�������3����ϡ��Һ�¶�Ϊ15�档����ϡ��ҺŨ�ȹ�����ͣ���ʹ������ճ�����Ż��ܽ⡣
���縴����Ȳŵ��ͪ��������[23]������Ȳŵ��ͪ(LNG)��ƶ���(Ez)���ȣ��ӵ�������Һ�л������ȣ�����������ҺΪ���ۼ��Ƴ��ң���״��ͼ6-7��������10-40µm��ռ������95�����ϣ�ƽ�������Ϊ20.7µm��
(3)��������
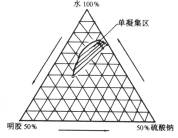
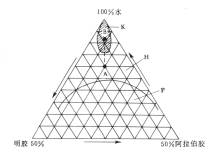
1) ����ϵͳ����ɣ������۷���������Ԫ��ͼ��Ѱ�ҳ���ϵͳ�������۵���ɷ�Χ��������-ˮ-������ϵͳ�ĵ�������Ԫ��ͼ(ͼ6-8)��
|
|
|
|
ͼ6-7 ��Ȳŵ��ͪ��ƶ�����������(��256��) |
ͼ6-8 ����ˮ�����Ƶ�������Ԫ��ͼ |
2)������Һ��Ũ�����¶ȣ�����������Ũ�ȿɼ��ٽ�����Ũ�Ƚ��͵�һ���̶ȾͲ��ܽ�����ͬһŨ��ʱ�¶����������������߹�ij�¶����ܽ�����Ũ�����ߵĿɽ������¶��������ߡ���5��������Һ��18�����²Ž�������15����������23�����½�����ͨ������Ӧ��37���������۳������ң�Ȼ���ڽϵ��¶���ճ����������������������۳���ʱ���¶���40��45��50��55��60��ʱ����ʡ�������С�ͷֲ�������ͬ����50��ʱ����Ϊ63%������65�����ϵ�������Ϊ5.5µm����40��45��ʱ�IJ��ʷֱ�Ϊ74����95%��������Ϊ5.5µm�ķֱ�ֻ��37.4%��33%����55��60��ʱ���ʷֱ�Ϊ72%��58%���Ҷ����ҵ�����С��2µm������CAP������ʱ����Na2SO4�����ۼ������Һ���������ˮ��Ľ��������ϴ����β��ã��������¶��Ҽ���ˮ�Խ��ͽ��������������Ը������Ρ�
3)ҩ�P�����������:�����۷���ˮ�Խ����г��ң����Ҫ��ҩ��������ˮ����Ҳ���ܹ�����ˮ��������γɲ���ҩ��Ŀ��ҡ�����ʱϵͳ���л����ܽ��ҩ��������ˮ���ࡣ�һ�������ȡ��������ͬҩ�������������ǿ���ױ��һ������ɽ�����������˵����
ƽ��ʱ�����ϼ��ֽ����������Ĺ�ϵ��ͼ6-9��
|
|
|
ͼ6-9 ����������ϵĽ���������Ӵ��� |
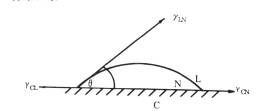
�����ʽ��: ��CL=��CN+��CNCOS��
6-1
ʽ6-1�ű�C��ʾҩ�L��ʾ��Һ��N��ʾ�����ࡣҪ��������ȫ��չ��ҩ���ϵ�������:�Ӵ�����=0��������CL����CN+��LN.ʵ���ϣ�ֻҪ��������ҩ�����൱����������ʹ
90����0��0����������Ҳ����ҩ���������ʪ����չ����ҩ�����ֲڲ������¶�ʱ�����ڽ����ܴ��������ճ�Ƚ��ͣ�����������ʪ��ҩ����������ǻ��һ�����ͣ��ɴٽ������ҵ��γɡ�
�����Ϊ�������ҩ�������ˮ���ױ�ˮ������ֻ������ˮ���ж����ܻ������������г��ң�����ۻ�轺�������ﶼ�������ˮ�����ܳ��ҡ���ҩ�������ˮ�����������к�������ˮ��ʹҩ��Ȳ��ܻ�����ˮ���У��ֲ��ܻ������������У�Ҳ���ܳ��ң���˫Ȳʧ̼����������˾��20���ʵ�����˫Ȳʧ̼������ˮ�ԣ��Ϳ��Գ��ҡ�ҩ�����ˮ���ʵ�������������ɰ��ң��������ɽ����80��ʹҩ����ˮ�Թ�����˲��ܳ��ҡ�
4)�����ҵ������Լ�����ˮ���Ľ�������:Ϊ�˵õ����õ������ң����ۺ��������Ӧ��һ���������ԡ�����A�������Ʊ���ʱ���ɵμ���������ʹ��Һ��pHֵ��3.2-3.8֮�䣬�ܵõ��Ȳ���pHֵ��С�������ң���Ϊ��ʱ�����������н϶��-NH3+���ӣ��������϶��ˮ���ӣ�����������-ˮ��Ľ��������������ҵ������Ժã�ʹ���������ڷ�ɢ��С���Ρ���������Һ��pHֵ��10-11���ܳ��ң���̫�ӽ��ȵ�㣬�д���ճ����״����������B��������pHֵҲ�ܳ��ҡ�
5)�̻������Ƶò�������ң��������̻����̻���ͬʱ��Ҫ���Ҽ��ճ���������á���ʹ�ü�ȩ���̻�����ͨ����ȩ���Ϸ�Ӧʹ�������ӻ��ཻ�����̻��������ij̶��ܼ�ȩ��Ũ�ȡ���Ӧʱ�䡢���ʵ�pHֵ�����ص�Ӱ�죬���������pHֵ��Χ��8-9������������������ճ�������������ȣ����������Ҵ���̫���䷴Ӧʽ����;
R-NH2+HCHO+NH2-R����R-NH-CH2-NH-R��+H2O
��ҩ�ﲻ���ڼ��Ի������ɸ������ȩ�����ȩ�������Խ���ʹ���������̻�.���ȩ�������Ĺ̻����ÿɲ���ϣ��Ӧ(Schiff reaction)��ʾ��
RNH2+OHC-(CH2)
3-CHO+H2NR����RN��CH-(CH2)
2-CH��NR��+2H2O
ʵ�������ȩ��ˮ��Һ�г��Ծۺ������ʽ���ڡ�������塢�����ȣ�����廹�����γɻ�״��
(4)Ӱ����ҵ�����
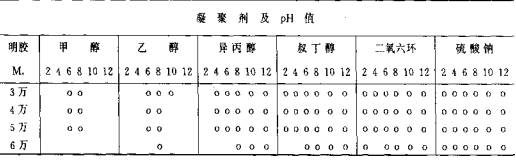
1)���ۼ��������pHֵ���õ���������ۼ�ʱ�������ӶԽ�������Ҫ���ã�ǿ������Ϊ�������ʯ��������Ȼ������廯��⻯������ӵ�������ߵĽ���������ǿ�����÷������ֱ�Ϊ3��4��5��6���A������(�ȵ��8.5)�����5����Һ����pHֵ�ֱ�ﵽ2��4��6��8��10��12ʱ�������롪����ҩ��ڽ����·ֱ����6�ֲ�ͬ�����ۼ��������ˮ�н��������á����룬���������ϴ����10����ȩ���������Һ�̻�����ˮ������ո�����ú�ҩ�ķ�ĩ״�ҡ��������6-4�����С�o����ʾ�����۳��ҡ����ü״������ۼ�ʱ����������(Mr)��3��-5���������pH 6-8�����۳��ң����������������ۼ�ʱ��M r��3��-6���������pH 2-12�������۳��ѣ������ơ�
��6-4 ��������������
2)ҩ�������������������õ����۷��Ʊ�����̿����������SD�ȼ���ҩ���������ʱ���ֱ����Ҵ��������Ƶ������ۼ���ҩ��������ɶ�����һ�������ƣ����������������������������ʹҩ���������ֵ���������������Ƶ�����ֵ�ϴ���(9-90mV)�������Ƶ������ң��������Ƶ�����ֵ��С��(0-8mv)�����������������ң�ֻ�е�ҩ��Ϊ����̿ʱ���ܰ������ҡ��о����֣������Ƶ�����ֵ��ӳ�˱���������������ʵ����������������Ҫ�ﵽһ���̶Ȳ��ܰ������ҡ�
3)���ܼ���Ӱ�죺Ϊ��ʹ�Ƶõ������Ҿ������õĿ����ԣ���ճ������ɢ�Ժã�����������ܼ�����ɽ�洼�����Ҷ���������������͵ȡ�Nikolayey���о��������ڵ����۷��Ʊ�������ʱ�������ܼ����ɼ����Ҿۼ��������ұں�ȣ��Ҽ������ܼ�����ͬ��ҩ��˥����1/2��ʸ���ء�
2�������۷� �����۷�(Complex coacervation)ϵʹ�����ִ��෴��ɵĸ߷��Ӳ�����Ϊ�����Ҳģ���һ�������½����������������۳��ҵķ������������ϲ��ϵ��������밢������(��CMC��CAP�ȶ���)������������������ᡢ����������Ǿ��ǡ�����������ס������밢�������ȡ�
���������밢������Ϊ����˵�������۷��Ļ���ԭ������������ҺpHֵ�Եȵ�����ϵ����ȵ������ʹ֮������(pH4.0-4.5)�������������Դ����磬���ڵ�ɻ������������γ����������ӵ������ܽ�Ƚ��Ͷ����۳��ҡ������۷��Ǿ�����һ���������������㣬�������գ��ʺ���������ҩ����һ��������۷��Ĺ����������£�
|
|
�����Һ��ҩ�� |
|
2.5-5%������2.5-5%����������Һ |
|
|||||||
|
|
�K�L |
|
|||||||||
|
|
�� |
|
|||||||||
|
|
����Һ����״Һ |
|
|||||||||
|
50-55�� |
�� |
5%������Һ |
|||||||||
|
|
������ |
|
|||||||||
|
30-40���ˮ |
�� |
����Ϊ����ϵͳ��l-3�� |
|||||||||
|
|
������ |
|
|||||||||
|
10������ |
�� |
37����ȩ��Һ(��20%NaOH��pH��8-9) |
|||||||||
|
|
�̻��� |
|
|||||||||
|
|
�� |
ˮϴ����ȩ |
|||||||||
|
|
�� |
|
|||||||||
|
|
�� |
|
|||||||||
|
|
�Ƽ� |
|
|||||||||
������������������Ϊ���ϣ�����ˮ�������������������ߵ��������������Ĺ�ϵ������ͼ6-10��Ԫ��ͼ˵����ͼ��KΪ���������������γ��ҵĵ�Ũ�������Ͱ������������Һ��PΪ�������������������������Һ���ܻ�������γ��ң�HΪ��������������Һ�ɻ����γɾ������Һ����A�����10��������10������������80��ˮ�Ļ��Һ�������ˮϡ�ͣ���A-B���߽���������K���ܷ������ۡ�
��ͼ˵��������ͬ������������������ʱ����pHֵΪ��Ҫ�����⣬Ũ��Ҳ����Ҫ������
���ȱ������������ң����߽�Һ̬�ȱ����������������������Ƴ���״Һ���Ӵ�����Һʹ�����۳��ͣ���ˮϡ�ͣ��ü�ȩ�̻����ã���ͼ6-11��
�����۷��������۷��Թ�̬��Һ̬��������ҩ����ܵõ�������ҡ���ҩ����涼���뱻�Ҳ�����������ʪ���Ӷ�ʹҩ��������黯�ڸ��������У��������������ɢ�����ҡ���˿ɸ���ҩ�������ʵ�������ʪ�������Ӧʹ�����ౣ��һ���������ԣ�������¶Ȼ��ˮϡ�͵ȣ����DZ�֤�������õı�Ҫ������
|
|
|
|
ͼ6-10 ����������������pH4.5��ˮϡ�͵ĸ�������Ԫ��ͼ |
ͼ6-11 �ȱ������������� (��254��) |
3.�ܼ�-���ܼ���
�ܼ�-���ܼ��� (solvent-nonsolvent)�����Ҳ���Һ�м���һ�ֶ��ҲIJ��ܵ��ܼ�(���ܼ�)����������룬����ҩ��������ҵķ����������Ҳĵ��ܼ��ͷ��ܼ�����ϼ���6-5��ҩ������ǹ�̬��Һ̬����������ܼ��ͷ��ܼ������ܽ⡣Ҳ����Ӧ��ʹ����ˮ�Ҳģ�Ҫ���л��ܼ��ܽ⣬��ˮ��ҩ������ҲĻ�ϣ���ˮ��ҩ�ﲻ�����л��ܼ����ɻ������黯���Ҳ���Һ�С�Ȼ����������л��ܼ��ķ��ܼ���ʹ���Ͻ����ܽ�ȶ�����Һ�з��룬�˹�����ȥ�л��ܼ������ҡ�
��6-5 �����Ҳĵ��ܼ������ܼ�
|
�Ҳ� |
�ܼ� |
���ܼ� |
|
�һ���ά�� �л���ά�� ������ά�ض��� ������ϩ ����ϩ �۴�����ϩ�� ����ϩ�����Ṳ���� |
���Ȼ�̼(��) ������ϩ ��ͪ �����(����) ���ױ� �ȷ� �Ҵ� |
ʯ���� ���� ����� ˮ(���Ҷ���) ������ �Ҵ� �������� |
���Ʊ��ٸ�ϸ���������ң����߽���ҩ���Ũ��Һ��ɢ��Һ״ʯ���У����黯����CAP(��ͪ���Ҵ��ܽ�)�����黯�������ȷ�(���ܼ�)���������ġ�������ϴ�ӡ�����������ɫ��ĩ״�ҡ�ƽ������12.7µm����ҩ��29.7%.ҩ��İ�����ʿɴ�95.7%��
4.�ı��¶ȷ� �����������ۼ�����ͨ�������¶ȳ��ҡ�EC���Ҳ�ʱ�������ڸ����ܽ⣬���³��ҡ�ʹ�þ��춡ϩ(PIB��Mmw=3.8��lO5)���ȶ����ɸ����Ҽ��ճ������PIB��EC����������ɵ���Ԫϵͳ����80���ܽ�ɾ�����Һ����������45������Ѹ������25����EC�����۳��ҡ�
�Ըı��¶ȷ���EC��ά����C�һ�ʱʹ���˼��ַ�ɢ��(Ũ�Ⱦ�Ϊ3%)����ֹճ����Ч���ǣ�������PIB��������ϩ�����հ�(���ӷ�ɢ��)������ҩ����PIB������ϩ���հף���������PIB����Ч�����������Ĥ�������������.����Mmw��ͬ�����в�ͬ����Mmw��3��6��105��6��105 ʱ�����������Χ�ֱ����˱���Ϊ4.7��-7����3%����Mmw��(2-4)��l05ʱ����3��������Ĥ���ҿɻ��͡�EC�ڻ������е��ܽ����FIB�ļ�������ͣ�����������������PIB��Mmw���������Mmw��PIB����75�������γ����ε�EC�ң�55����65����ת��ɲ������Σ�����ճ�������ø�Mmw��PIBʱ���γɵ���������С���ʷ������״ʵ�塣���������Ʒ����ڻ��������Ƶ�ά����C��EC�ң���һ���Ļ����Բ���ֹ��������������ҩ����һ������ѧ����.����ҩ��������Ĥ���������ݼ���Ҳ�����÷������Ҳģ���ά����C�������ȱ�ͪ�г��ң���ȴ�̻����ҡ�
5��Һ�и�� ����״Һ�г�ȥ��ɢ��ӷ����ܼ����Ʊ��ҵķ�����ΪҺ�и��(in-liquid drying)�������˳�Ϊ�ܼ��ӷ�����
Һ�и���ĸ��﹤�հ��������������̣��ܼ���ȡ����(��Һ��֮��)���ܼ���������(Һ�������֮��)���������ɷ�Ϊ�����������Ъ������鷨��ǰ����Ӧ��O/W�͡�W/O�ͼ�O��O��(�����棯Һ״ʯ������ͪ��Һ״ʯ����)��״Һ�����鷨Ӧ��W/O/W�ͻ�O/W/O���顣���Ƕ�Ҫ���Ʊ��Ҳĵ���Һ���黯���Ҳ���Һ������״Һ�еķ�ɢ�࣬����������ܣ����Ҳ��ܼ���������Ӧ��һ�����ܽ�ȣ�������ȡ������ʵ�֡������������Ъ����У������õ��Ҳ��ܼ������ܽ�ҩ����Ƶõ�������õ������ң����鷨�Ƶõ����ҡ�
��������Ʊ��ҵĻ��������������£�
|
���ӷ��ܼ��н��Ҳ��ܽⲢ��ҩ���ɢ |
|
||||||
|
|
�� |
�������༰�黯�� |
|||||
|
|
����Һ |
|
|||||
|
|
�� |
����������ȥ�Ҳĵ��ܼ� |
|||||
|
|
�� |
|
|||||
���Ҳĵ��ܼ���ˮ�����ܣ�����ˮ�������࣬������ˮ���黯��(�缫�ԵĶ�Ԫ��)���Ƴ�O/W����״Һ������ø߷е�ķǼ���Һ����Һ״ʯ���������࣬�Ƴ�O/O����״Һ�����Ҳĵ��ܼ�����ˮ���ܣ������������Һ״ʯ���������������黯��(��˾��80��85)���Ƴ�W/O����״Һ����������������IJ�ͬ���ֿɷֱ��Ϊˮ�и�������и����
�粼��Ҽȿɲ���ˮ�и������ɲ������и���Ʊ��ҡ�ˮ�и���һ��IJ�������EC����CH2Cl2�С������100Ŀɸ�IJ���ҷ�ĩ����30��ˮԡ��250RPM����20���ӣ��������裬���뺬0.5��������Լ���100m1����ˮ�У�ˮ����30�������ߵ�40�棬330RPM����3Сʱ���˹�����50m1����ˮϴ��3�Σ����¸���24Сʱ�����÷�ĩ״�ҡ����и���һ��IJ�������Eugragit RS���ڱ�ͪ�У������100Ŀɸ�IJ���ҷ�ĩ����10��ˮԡ��250RPM����20���ӣ��������裬�ӵ���190RPM�����µ�ͬһˮԡ�е�Һ״ʯ��200m1�У�ˮԡ�¶���10�������ߵ�35�棬��190RPM�½���4Сʱ���˹�����������ϴ��3�Σ���ѹ���T�÷�ĩ״�ҡ�
��O/W������Һ����������������ұ��泣��ҩ������塣��������Ƹ������ʣ�ʹ����������һ���ˮѸ����ȡ�γ�ӲĤ�ټ���������ɵ�������ң��Ƽ�Ъ�����
����������Ъ�������ˮ�������࣬��������ˮ����ҩ����ң������е�ҩ������ˮ������Ͱ�����ʺ���ҩ�����ɲ���ˮ������O/O������Һ��
��O/O������Һҩ���Կ������������γ����壬����ҩ������������Ҳ��������Ժõķ�ĩ��
���鷨�ɿ˷�����ȱ�㡣����W/O/W���鷨�Ĺ����������£�
|
�Ҳĵ��л��ܼ���Һ(���������黯��) |
|
ҩ��ˮ��Һ(��������) |
|
|||||
|
|
�K�L |
|
||||||
|
|
�� |
|
||||||
|
|
W/O������Һ |
|
||||||
|
��ȴ(15��)������ˮ��ճ�� |
�� |
�Ӻ���ˮ���黯����ˮ�������� |
||||||
|
|
W/O/W���� |
|
||||||
|
|
�� |
������ȥ���ϵ��ܼ������롢���� |
||||||
|
|
�� |
|
||||||
����������ECΪ�Ҳģ��Ը��鷨�Ʊ���ʱ���ɽ���������ˮ��Һ��ɢ�ں�EC�����������л������γ�W/O������Һ������������EC�ڷ�ɢ���������Ľ���ֱ��γ���������Ĥ����ʾ��ͼ6-12(a)������Һ��һ���밢��������Һ�黯���γ�W/O/W���飬�����µ�ˮ/�ͽ��棬����������EC��һ���γ���������Ĥ����ͼ6-12 (b)������ȥ�ڡ���ECĤ֮������������л��ܼ����˹���������㶼�ǰ�������Ĥ���м���ECĤ������Ĥ���ң���������50µm���£���ͼ6-12(C)��
|
|
|
ͼ6-12 W/O/W����ʾ��ͼ |

(��)�������
�����ǽ���̬��Һ̬ҩ���������н����һ�����Ҫһ���豸������
1��������� �������(spray drying)�ֳ�Һ�����������������������ڹ�̬��Һ̬ҩ����һ���������ΧΪ5-600µm���������Ƚ��������ɢ���Ҳĵ���Һ�У��������������˻�����������������ʹҺ�����������Σ���������̻����������ﲻ�����Ҳ���Һ���ɵõ��ң��������Ƶý��ζ�����ѹ�ĺ����������ң���ͼ6-13��
|
|
|
ͼ6-13 �����Ѽ���������������� (��15000��) |

�����ܽ⣬�ɵ����ܽ��Ҳĵ��ܼ�������ˮ��Ҳ�������л��ܼ���Ŀǰ��Ҫ��ˮ���ܼ������������豸�μ������¡���������װ�á���
�����ɲٷ��Ĺ���Ӱ�����ذ������Һ��ճ�ȡ������ԡ�ҩ�P�Ҳĵ�Ũ�ȡ����������ʡ������������������ʵȡ����������ɻ��ҺŨ��������ڵ��¶Ⱦ����������������������̫����ܱ���Ĥ����.ͨ����Ĥ��ף��������Ҳ�Ʒ���ܶȽ�С.��������ΪҺ̬��ͨ���������к���������30%.
�Ҵ���������ճ������������θ������.���Ҳ��к��о��Ҷ�������ճ��ʱ���ɽ����Ҵ����������ճ��;������ʹ��ˮ��ˮ��Һ.���ڹ����в���������������Ъʱ�����ɼ����Ҵ��������ճ��.����С�����������ʱ���Ҳ���Һ�м��뿹ճ���Ƴɻ���Һ���ɼ�����ճ�������õĿ�ճ������6-6���������裬��ʯ��Ӳ֬��þ������Է�״����Ͳ��Ʒ�У��Լ�������ʱ��ճ��������ѹƬ��װ���Ľ���ʱ�����ҵ������ԡ�
��6-6 ����ʱʹ�õĿ�ճ��������
|
��ճ�� |
�����Ҳ���Һ�� (g/100g�Ҳ�) |
�����ҳ�Ʒ�� (g/100g) |
|
��ʯ |
20-100 |
1-3 |
|
�轺 |
3-20 |
1-3 |
|
Ӳ֬��þ |
10-50 |
0.5-3 |
|
��Ӳ֬������� |
1-3 |
- |
�Ʊ��Ź��������Ӱ�������ϰ�(gadopentetate dimeglumine)��EC��ʱ�����ö��(5��)�������·�����Ʒ����126µm[24]��
2. �������ᷨ ���������ɢ�����ڵ��Ҳ��У��������������������۶����ҵķ�������Ϊ�������ᷨ(spray
congealing)�������ֵ��Ҳ������ࡢ֬�����֬�����ȣ����������¾�Ϊ���壬���ڽϸ��¶������ڣ�������������(mexiletine hydrochloride)Ϊ�������Ӳ֬���ECΪ�����Ҳģ���34.31-68.62kPa��ѹ������ͨ���������ᷨ���ң�����8-100µm��
3. ���������� ����������(air suspension)������������·�(fluidized bed coating)��ϵ����ǿ����ʹ�����������ڰ���
���У��Ҳ���Һͨ��������������������棬ʹ����������������������Һ�Ӹɣ������������γ��Ҳı�Ĥ����.�豸װ�û�������Ƭ����������װ����ͬ.�������õ�������һ����35-5000µm��Χ.�ҲĿ����Ƕ���ǡ���������֬��������ά�������P�ϳɾۺ���.���������ҵĹ����У�ҩ�������ۻ����������������¹����п��ܻ�ճ�ᣬ��˿ɼ�������ֳɷ��绬ʯ�ۻ�Ӳ֬��þ�������ۻ�ҩ��ճ���һ����λ��Ȼ����ͨ�����������£��ɿ˷��ۻ�ҩ���ճ�ᡣ
�����������ٱ���Ĥ�������¡�����Ĥ�ȼ��������ܿ����ͷŵ�Ĥ��������ˮ��Һ���������������·��Ʊ���
4��������ķ� ����������ʹ��������ٴ����Ҳĵ�Һ̬Ĥ���ٽ���̻�ԡ�̻��Ʊ��ҵķ�����Ϊ������ķ�(multiorifice centrifugal process)��������ԲͲ�ĸ�����ת���������������õ����Ӳ�������Ҳ�ԡҺ�γ�Һ̬Ĥ��������(Һ̬���̬)���ٴ���Һ̬Ĥ�γ��ң��پ�����ͬ�������Թ̻�(�÷��ܼ���������ȥ�ܼ���)�������ҡ�
5�������·� �����·�(pan coating)ϵ���ð��¹����Ҳ���Һ���ڹ�̬�������ϻӸ��ܼ��γ��ң�������¹����������ɼ����ܼ��ӷ���
��������������е����������ˮ���Ժ�֬���Եġ���̬��Һ̬ҩ����һ������������������á�ͨ��������������е��ʱ��������һ����ʧ������ճ��������������ʧ��5�����ҡ�ճ����10�����ҡ������ж���Ϊ�Ǻ����ġ�
(��)��ѧ��
��������Һ�е����߷���ͨ���ۺϷ�Ӧ�����Ϸ�Ӧ��������Ĥ�Ƴ��ң������һ��ķ�����Ϊ��ѧ�����������ص��Dz������ۼ��������Ƴ�W/O������Һ�������û�ѧ��Ӧ�����̻���
1���������۷� �������۷�(Interface po1ycondensation)��ƽ���ۺϷ������ڷ�ɢ��(ˮ��)��������(�л���)�Ľ����Ϸ�����������۷�Ӧ�����磬ˮ���к���1��6-�Ѷ����ͼ�л���Ϊ���Ա��������ȵĻ����顢�ȷ���Һ�������������Ͻ��裬��ˮ�ν����Ϸ������۷�Ӧ�����ɾ���������Ӧʽ���£�
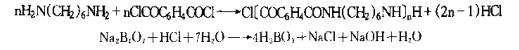
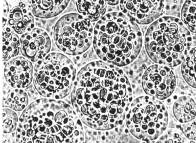
�������۷�Ӧ�����ʳ���l��6-���������л�����ɢ�����ʣ��ʷ�Ӧ���ɵľ�����������ȫ�����ڽ����Ϊ�Ҳġ���L-�Ŷ�����ø�ң�ȡL-�Ŷ�����ø10mg���Ŷ���50mg����1m1����O��Ѫ�쵰��Һ��1.5m1��pH8.4���Ỻ����Һ�У���1ml 1��6-�������ļ�����������Һ�����ڷ�Ӧƿ�У��ټ�20m1����Լ�(�ɻ�����150m1���ȷ�30m1��˾��85 0.9ml�������)����4���ԡ��3000RPM����1���ӣ��ӶԱ���������15mL����������5���ӣ�����30m1����ܼ�����������0.5���ӣ������¹۲����γ��ҡ���������ת�����Ĺ��У�1000RPM����1���ӣ�ȥ����Һ����25m1��ɢҺ(12.5m1����20��12.5m1����ˮ)������3���ӣ���50m1����ˮ����������1���ӣ�����ȥ����Һ���һ�����������ˮ�У�4�汣�档��ƽ������20µm����ʾ��ͼ��ͼ6-14��
|
|
|
ͼ6-14 �Ŷ�����ø�Խ������۷�����ʾ��ͼ |
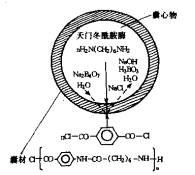
2�����佻���� �÷�ϵ���������黯״̬�£������������䷢���������ٴ����Ƶ÷�ĩ״�ҡ��ù��յ��ص��ǹ��ռ��������������������ɷ֡������Ŷ�����ø����Ϊ���������乤���������£�
|
50g/L����ˮ��Һ |
|
Һ״ʯ��(���黯��Ӳ֬���) |
||||||||||||||
|
|
�K�L |
|
||||||||||||||
|
|
�� |
����30���� |
||||||||||||||
|
|
�ȶ�����Һ(W/O��) |
|
||||||||||||||
|
|
�� |
ͨ���� |
||||||||||||||
|
|
��������Һ |
|
||||||||||||||
|
|
�� |
60Co����Դ����(������Ϊ25. 8C��(kg��h)����������774-1290C/kg) |
||||||||||||||
|
|
����Һ |
|
||||||||||||||
|
|
�� |
���������飬��ȥҺ״ʯ�� |
||||||||||||||
|
|
������ |
|
||||||||||||||
|
|
�� |
������ϴ���Ҵ�ϴ����ո��� |
||||||||||||||
|
|
��ĩ״������ |
|
||||||||||||||
|
|
�� |
�����Ŷ�����øˮ��Һ����������ˮ |
||||||||||||||
|
|
��ĩ״�Ŷ�����ø������ |
|
||||||||||||||
���Ƶõ������������ɴ�30µm���£�����0.25��1.0��10.5��47��72Сʱ��������ҩ��(������λ)�ֱ�Ϊ202��566��739��769��789������ҩ���ɷ��Ϸ���3/2[1-(1-Q)2/3]-Q��f(t)��
l-�Ŷ�����������ϸ���ı���Ʒ������ϸ���������ϳɣ�����ϸ�����뿿�����ṩ�����Ŷ�����ø��1-�Ŷ�����ˮ�⣬��ϸ��ʧȥ����������l-�Ŷ��������������Ŷ�����ø���������Ƽ����ܰ�ϸ����Ѫ������ø���������Ե��ף������������Ӧ������ע��ɲ��������ʧ������Ʊ����ҿ��Ա��������Ӧ��ʧ����˽��Ŷ�����ø�Ҷ�С����ǻע�䣬���ֵ�һ�ֲܷ���������ǻ�ڣ��ڶ��ܸ�ǻ�ϲ�������Ũ����һ��ע��70��λ���ڸ�ǻ����Ч�����ٱ���4�ܡ�һ���Ŷ�����øˮ��Һע�����Ϊ���Ե�����Ѹ����й�����ڵ��Ŷ�����ø�������ڸ�ǻ�ڣ������ͷ�ҩ�������������������
�����ҵ�����
(һ)�ҵ���̬���ṹ������
1���ҵ���̬��ṹ �������ӦΪ��С���ȵ����Σ�������֮�䲻ճ������ɢ�Ժã������Ƴ��Ƽ����ҵĽṹ���Ź��������IJ�ͬ���в��졣ͨ�����������۷�����佻�������õ�����������Ƕ�ͣ����Ƕ������������ɢ��Ƕ���������ڡ�������е�����������۷����ܼ�-���ܼ����Լ�������ҷ����õ���������Ĥ���ͣ�����������е���Ƶõ��ҿ��Ժ������������������������۷�ֻ���Ƶõ��������ҡ�
�ұ���Ӧ����һ���Ŀ����Ժ͵��ԡ���������Ϊ�Ҳģ�����l0��-20�����ͻ���������ɸ��������Ҳĵĵ��ԣ��������ճ���һ���ά�ؿɼ���Ĥ�ڵ�ϸ�ף���ɼ���70���ǽ�ʹ����Ե�ȱ��õ����ƣ������һ���ά��Ϊ�Ҳģ�Ӧ�������ܼ������������.
2���ҵ����� �ڷ�����С��200µm����(��ճ�Ե�Һ���ʳ�ﹲ��)ʱ���ڿ�ǻ�ڼ�������С��ʳ�Ҫ���Ʊ�С�����Ŀڷ��ҡ�������̫С�����ʱճ�����ء��ҵ�������ֱ��Ӱ��ҩ����ͷš��������öȡ���ҩ�����л��ܼ��������Լ����ڷֲ��İ����Եȡ�
��25��Ȳŵͪ�Ա������ҽ���(85��15)������Ϊ�Ҳĵ�����37�桢275g/L�Ҵ�Һ��һ��ʱ���ڵ������ͷ������������ҵ�������������͡�������ܼ�-���ܼ����Ƶ�˳���ң��ڲⶨ���л��ܼ����ȼ���IJ�����ʱ����������20-50µm�IJ�����Ϊ0.028����100-300µm��Ϊ0.191����200-300µm��Ϊ0.760����300-400Pm��Ϊ1.830����˵�����������л��ܼ����������ࡣ
12-19µm��˿��ø��C�Ҿ�ע�������ڼ��зֲ��ڷΣ��߷�ʱΪ95.4����2СʱΪ88.4����24СʱΪ59.9�������ڸ���ֻ��3.6��������142Ce�۱���ϩ����ϩ�����ң���Beag1eȮ��ע��7-12µm����������24Сʱ�����ڷ��ڴ�42.8��-58.8�������ڸ���ֻ��6.5��-0������3-5µm���Ҽ����ڷε�ֻ��1.2��-12.4�������ڸ�����62.5��-50.9����
(��)Ӱ��������������
1. ������Ĵ�С 150-250µm��ά����C��ĩ�����
�뷨�Ƴ��ң���ƽ������Ϊ512µm����������Χ�ֲ���խ��С��120µm��ά����C�Ƶõ���ҲС������������Χ�ֲ��Ϲ㣬Ϊ250-500µm.�ɸ��ֵ�ά����C���Ƴɵ��ҽϴֲ���ΧҲ�㡣ͨ����Ҫ���ҵ�����ԼΪ10µmʱ������������Ӧ�ﵽl-2µm��Ҫ���ҵ�����ԼΪ50µmʱ������������Ӧ��6µm���¡��Բ�����ˮ��Һ̬ҩ�������뷨�Ʊ���ʱ�������黯���һ����ɵ�С�����ȵ��ҡ�
2. �Ҳĵĵ����� һ��ҩ��������С����������Ҫ�Ƴ��ұں����ͬ���ң������Ҳ�����.
3. �Ʊ����� �Ʊ�����Ӱ���ҵ�����������6-7.
4. �Ʊ��¶� ���һ���ά��Ϊ�ҲĵIJ���ң����������Ҳĵ�������Ϊ1�U1���ױ�-ʯ����Ϊl�U4�������ܼ�-���ܼ�����������380RPM�����¶ȷֱ���0��20��40��������������6-8.
��6-7 �һ��������������Ժ�������Χ
|
�һ����� |
���õ������� |
������Χ(µm) |
|
�������� |
��̬ҩ�� |
35-5000* |
|
����� |
��̬��Һ̬ҩ�� |
2-5000* |
|
������� |
��̬��Һ̬ҩ�� |
1-5000* |
|
������ |
��̬ҩ�� |
5-5000* |
|
������������� |
��̬��Һ̬ҩ�� |
5-600 |
*���������ɳ���5000µm.
��6-8 �¶ȶԲ����������Ӱ��
|
������(µm) |
��90 |
��150 |
��180 |
��250 |
��350 |
��425 |
��710 |
��1000 |
|
|
��ͬ�¶� �µ��� ����(%) |
0�� |
12.0 |
49.8 |
95.8 |
97.8 |
98.3 |
99.1 |
99.9 |
- |
|
20�� |
2.2 |
15.7 |
42.1 |
84.7 |
73.1 |
77.9 |
91.4 |
94.8 |
|
|
40�� |
0.5 |
3.9 |
10.3 |
62.0 |
76.3 |
89.2 |
93.9 |
98.4 |
|
5���Ʊ�ʱ�Ľ������� ��һ���¶��¸��ٽ��裬������С�����ٽ���������Ѫ�쵰������800RPMʱ����ƽ������Ϊ19.2µm���������Ȼ�������ת�ٸߣ���ƽ������Ϊ4.9µm���ҡ�
�������Ƶ���߽����ٶȣ��ҿ�������ײ�ϲ�����������⣮����������ȡ���ڹ��յ���Ҫ��������Ϊ�Ҳ�ʱ��������뷨�Ʊ��ҵĽ������ʲ���̫�ߣ�����������ԼΪ50-80µm������ٽ�������������ݻή���ҵIJ�����������
6�����Ӽ���Ũ�� ������ý������۷��ҽ�������һ�£����ֱ����Ũ��Ϊ0.5����5%��˾��85��ǰ�߿ɵ�С��100µm���ң����������С��20µm���ҡ������ñ������ҽ���(������78��22)������Ϊ�Ҳģ��Ʊ���Ȳŵͪ���ʱ�������黯��������Ũ�Ȳ�ͬ��ƽ��������ͬ��1��������70.98µm��2��������79.8lµm��3��������59.36µm��4��������46.77µm��
(��)����ҩ����ͷ�
ҩ���һ���һ��Ҫ��ҩ���ܶ�ʱ�����ش������ͷų������ﵽ�ٴ�Ԥ����Ҫ��
1.����ҩ���ͷŵ���������� Luu Si-Nang�ȶ�����������ҩ����ͷ������������۴�����������ͷ�����Ϊ��
(dc��dt)��=(3Dm��Vrd)(Nh��a+����h)(C1-C) 6-2
ʽ��D��ҩ����ӵ���ɢϵ����m���ҵ���������V�ǽ��ʵ��ݻ���r���ҵ�ƽ���뾶��d���ҵ��ܶȣ�N�ǽ������ʣ�a��b�dz����� �������ұڵĶ�����йص����Գ�����h���ұڵĺ��(��������Ƭֱ�Ӳ�û�������������Ҳĵ�����������ֱ�����й���)��C1��ҩ����ܽ�ȣ�C��ҩ���ڽ����е�Ũ�ȡ���ʽ6-2��֪����һ����ҩ����Ҳ���˵������ҩ���ͷŵ�������������h��r���йء�����ʽ���֣���C1����Cʱ�ã�
C��C1(3Dm��Vrd)(Nh��a+��/h)t 6-3
��ʽ��ӳ�Ĺ������㼶�ͷţ������й���������ʱ������Ũ����ʱ������ȣ��༴�ͷ�����Ϊ����������ʱ��ı䡣�о�������ˮ�������ҡ��л����ͷ�������ҡ��������ҵ�ȷ����ˡ���������Ϊ�Ҳĵ��ȱ����������ұڽϱ�ʱ�����������㼶�ͷŹ��ɣ������ұ����ӵ�10.4µm����ʱ��C��t֮��ų������ȹ�ϵ��
����ҩ����ͷ�Ҳ�з��������ͷŹ��ɵġ�������������������CAP�Һʹƶ��������ң������ͷŷ���Higuchi���̣������ȴ����һ���ͷŹ��ɡ�
Ҫ�����˽�����ҩ���ʹ��ɣ�Ӧ������ҩ���ƣ�ͨ�����������֣�
(1)��ɢ��ҩ�����ұ���ɢ�����ҽ������ں���Һ������������ʹ����ҩ���ܽ���ɢ���ұڣ������������̣��ұڲ��ܽ⡣Ҳ�������ҩ���ͷ������Ǽ��ܽ��ճ�����ұ��е�����ҩ������ݵĿ����ͷţ���Ϊͻ��ЧӦ(burst effect)��Ȼ������������ܽ�ɱ�����Һ����ɢ���ҡ������ȱ������ҵ��ұڽϺ�ʱ��ҩ����ͷſɷ�Ϊ4���Σ��ٳ��ڵ�Ѹ���ͷţ������ܽ����ұ��е�ҩ��������ͷš���������ҩ����ܽⲢ��ɢ���ұڣ��۽Ͽ��ٵ���̬�ͷţ���������ҩ��ı�����Һ��ά��ʱ��Ҳ��������ϻ������ͷţ�����ҩ��IJ������֡���ʱ�Ѳ�����ά�������Ũ���ݶȡ�
(2)�ұڵ��ܽ⣺�ұ��ܽ��������Ҫȡ�����Ҳĵ����ʡ���Һ���������ɡ�pHֵ�Լ��¶ȵȣ���������ø�����á�����������ѧ���̡�
(3)�ұڵ������뽵�⣺������ø�����µ��������̡����ҽ������ں��ұڿ���θ����ø������ø�������뽵���Ϊ���ڵĴ�л�����ʹҩ���ͷų����������úϳɵ����オ��ľۺ������Ҳģ��ڽ���֮ǰ��ҩ�����ѿ�ʼ�ͷš�����������ҽ���[50�U50]������Ϊ�Ҳĵ��ɸ��Ĵ�����(naferelin acetate)���������ͷ���������ͷŻ��Ʒ�3���Σ���һ��Ϊ����Ŀ����ͷŽΣ�ҩ����ұ�����ɢ�ͳ����ڶ����Ǿۺ���ˮ�Ⲣͬʱ��������С�����Ա����䲻���ԣ�ҩ����ɢ�ͳ����������ǵͷ�����Ƭ���ܽ�;ۺ����������ʴʹҩ���ͷš��������������Ϊ�ұڵĽ��⡢�������ܽ⣮��ҩ�����뾭���ܽ�����ɢ�����ֲ�ͬ���ͷ����ʡ���˲��ܽ���ȫ�����á���ֱ�߱�ʾΪ�㼶�ͷš�
2.Ӱ����ҩ���ͷ����ʵ�����
(1)�ҵ����������ұڵIJ��Ϻͺ����ͬ�������£���������С�����������ҩ����ҲӦ��������ǰ�����ң����ۻ��ͷ�������������С�����ߡ�
(2)�ұڵĺ�ȣ��ұڲ�����ͬʱ���ұ�������ҩ������Ҳ����˵�����������ұڵ���������С����ҩ����������ǰ������ң��һ���ά��Ϊ�Ҳġ��ҵ��ұں�ȷֱ�Ϊ5.04��13.07��20.12µm�����˹�θҺ���������ܳ����ʲⶨ�������t1/2��ʾ���ֱ�Ϊ11��16��30���ӣ������ֱ�߷���t1/2��1.17h+4.32��˵���ҵ��ұ�(h)�����ͷ��������͡�
(3)�ұڵ�������ѧ���ʣ���ͬ���Ҳ��γɵ��ұھ��в�ͬ��������ѧ���ʡ����������γɵ��ұھ�����״�ṹ��ҩ��Ƕ����״��϶�У���϶�ܴ�ʽ6-2�еij����������ҩ���ܽϿ����ͷš������ұ��ɾ������γɣ����϶�뾶С(Լ1.6nm)��ҩ���ͷű����������öࡣ��϶�ʽ�С�����Գ�������С���Ҳģ��γɵ�����ҩ���������õļ����Ҳ��γɵ��ұ���ҩ���ʵĴ������£�
�������һ���ά�أ�����ϩ-�����������������
���ý������۷����ң���ˮ���ڼ�����ҳ�����(�����ҳ��İ�)�������������γɾ������ұڿ�϶������ʹ��ҩ�ӿ졣������-�������������۷����ң������ӻ��ͷţ��ɼ���������ճ��(0.4cPa��s)���һ���ά�أ�ʹ��������ұڿ�϶�ڶ������ֿ�϶��������ҩ���ʡ�
�����Ҳ����в�ͬ����ҩ���ʡ���ǰ������һ�ʱ��������-�����γɵ��ұ���ҩ����������-�������Ƶ��ұ���ҩ��졣����������ң����ұ�ȫ���һ���ά����ɣ�30������ҩ������70�������ұ��к�10��-30���ľ��Ҷ���ʱ��30���ӿ��ͷ�90�����ϵ�ҩ�����ѹƬʱ�������Ҷ���������ѹ����������ʹ���Ҷ������ڣ����̻���ʹƬ�����������ҩ����Ҳ��֮���͡�
(4)ҩ������ʣ�ҩ����ܽ����ҩ���ͷ����������й�ϵ�����Ҳĵ�������ͬʱ���ܽ�ȴ��ҩ���ͷŽϿ졣�������һ���ά��Ϊ�Ҳģ��ֱ��Ƴɰͱ����ơ������ἰˮ�����ҡ�������ҩ����37��ˮ���ܽ�ȷֱ�Ϊ255��9��0.63g/L���ͱ����Ƶ��ܽ�����ҩ����ͷ�����Ҳ���ǰͱ��������ҩ�����ұ���ˮ֮�����ϵ���Ĵ�СҲ��ӳ��ˮ�е��ܽ�ȴ�С������Ӱ���ͷ����ʡ����Ҳ�Ϊ�һ���ά�صİͱ����ơ������ἰˮ�����ң����һ���ά�أ�ˮ�ķ���ϵ���ֱ�Ϊ0.67��58��151�������ҩt1/2�ֱ�Ϊ22��70��80���ӣ������ͱ������ͷ���졣
���ʹҩ�ﻺ�͵ķ���֮һ���ǽ�ҩ�����Ƴ��ܽ�Ƚ�С��������������ɢ�壬Ȼ���ٽ����һ���
(5)���Ӽ���Ӱ�죺Ϊ��ʹҩ���ӻ��ͷţ��ɼ�����ˮ��������Ӳ֬�ᡢ������ʮ�����Լ�����������ȡ������������һ�����������ڷ����Ƶõ��ң��ڷ���8-10Сʱ���ܾ����ͷŵ�����������ֹ���ϩ����(metrazol)�Դ����շ����ʿɴ�10Сʱ֮�á�����ǰ�����һ���ά���Ҳ��ò�ͬ����Ӳ֬��Ϊ���ͼ����������ͼ��������ӣ�ҩ�������ͷ����ʽ��͡���Ӳ֬�������л��ܼ��У����Һ�������һ���ά���ұ������������ã�����Ĥ��ʹĤ����֮�ʡ�
(6)������������ͣ�����ʱ���������������ͬ��������������ͬ������ҩ����Ҳ����ͬ������������������������ң�����ҩ���ʱȺ���������Ҫ��Щ����������ں���ÿ���������������������ƽ����ǰ���߶�ö࣮���������С�������ҩ���������罫�������������Դ�����ά�ض�����Ϊ�Ҳģ����ò�ͬ���黯���գ����õ�����8Сʱ����ҩ���������6-9��
��6-9 �һ����黯������ͬ���ͷ�ҩ���Ӱ��
|
����* |
������(µm) |
8Сʱ����ҩ��(%) |
����ҩ�ﺬ��(%) |
|
a |
769 |
33��5.6 |
77��1.3 |
|
b |
659 |
40��1.9 |
83��1.8 |
|
c |
783 |
51��4.1 |
36��2.9 |
*a.�黯-�ܼ�����; b.�����黯-�ܼ�;c.�黯�������ܼ�
˵�����������С��࣬�����黯-�ܼ������Ƴɵ��ң���8Сʱ���ͷŵ�ҩ������������������黯�������ҡ�
ͨ������һ���Ļ������ã��������谷��������Ƭ��������ܳ�������t1/2��ʾ��ǰ��Ϊ8���ӣ�����Ϊ37���ӡ�����������Ƭ����ȣ����ߵ���ҩ���ܽϿ죮��ѹƬ���ұڿ��ܱ䱡�����ѡ������һ���ά��Ϊ�Ҳĵı��ͱ���������Ƭ����ȣ�������ҩ�Ͽ졣�����Һͷ��ø��������ҩ����Ҳ���ܲ�ͬ�������pH l.4��������Һ����ҩ�����ʵıȸ������Ī��ƽ����ҩ���ʴ�Լ��1������Ϊ�������Ҫ�Ⱦ�����ˮ���Ͳ�����Ч����ҩ��
(7)pHֵ��Ӱ�죺�ڲ�ͬpHֵ�������ҵ���ҩ����Ҳ���ܲ�ͬ�����ԿǾ���-��������Ϊ�Ҳĵ���Ī��ƽ�ң��ֱ���pH l.4��pH 7.2�Ļ�������Һ�вⶨ����ҩ���ʡ���pH 7.2ʱ����ҩ���Կ���pH l.4ʱ�����������Ҳ��еĺ���������pH�ϸ�ʱ�ɻ����ܽ����������ѡ������������������-��������Ϊ�Ҳģ����������������pH2ʱ������40�����ͷ�����ິ�100������pH9ʱ��120������ҩ������80����
(8)�ܳ���������ǿ�ȵ�Ӱ�죺�罫ӫ����������50mg������4L pH 7.4������ǿ��Ϊ0.8��1.0��1.2�������λ�����Һ�У���1Сʱ������ҩ����ֱ�Ϊ38.78����64.35����71.99����˵����ͬ����ǿ�ȵ���ͬ���ʣ����ͷ�ӫ���ص�����Ҳ��ͬ��
�ġ�������������
�ҵ����������DZ�֤����ҩ���Ӧ�����õ���Ҫһ����Ŀǰ������Ʒ���Ƽ��������Բ���������ҵ�����Ҳ����Ҫ����������о���
Ŀǰ�ҵ��������ۣ����Ƴ��Ƽ�Ӧ����ҩ���й��Ƽ��Ĺ涨�⡢���»�������������.
(һ)�ҵ�����������
�ҿɲ��ù�ѧ������ɨ�����������۲���̬���ṩ��Ƭ������̬ӦΪԲ�����λ���Բ�εķ����״�
��ͬ���Ƽ����������в�ͬ��Ҫ��ע�����������Ӧ����ҩ���л���ע����Ĺ涨�����ھ���ע�����������ʱ��Ӧ���Ͼ���ע��Ĺ涨��Ӧ�ṩ����ƽ��ֵ����ֲ����ݻ�ͼ��(��ֱ��ͼ��ֲ�����ͼ)��
�������IJⶨ�ж��ַ���������У�����Ĵ�Ŀ�����ǵĹ�ѧ�����ⶨ��ȡ�������ز�Ƭ�ϣ���Ҫʱ�ø��ͻ�Һ״ʯ��ϡ��(1-2)��ֱ�ӹ۲�����500������������������Χ����Ϊ���ɵ�Ԫ(��5-10��10-15��15-20µm��)������ʽ6-4������ƽ����DRV����ʽ6-5���ÿ��������Ԫ������ռ�������ٷ��ʣ�
![]()
ʽ��n1��n2������nnΪ��������d1��d2����dn����������gΪijö����Χ����ռ�����ٷ��ʣ�NΪ��������ij��������Χ��������dΪij������Χ�ڵ���ƽ��������
������õ��������ݣ�������Ϊ�����꣬��Ƶ��Ϊ�����꣮�ɻ���ֱ��ͼ��Ƶ�ʿ������Ұٷ��ʱ�ʾ(ÿһ��Ԫ�������Ҹ�������������)��Ҳ�����ø���Ԫ�������ٷ��ʱ�ʾ����ʽ6-5��
�����ֲ�����ÿ��(span)�����������С�ֲ���խ�����������ȣ�
��ࣽ(D90һD10)��D50 6-6
ʽ��D10��D50��D90�ֱ��ʾ��10����50����90���ҵ�������С�ڸ�ֵ��������
����õ��Ӧ��(��Coulter������)����Ӧ��(�����ȷֲ���Ȳⶨ��)�ⶨ�ҵ���������ֲ���
(��)����ҩ�ﺬ���IJⶨ
����ҩ�ﺬ���IJⶨһ������ܼ���ȡ�����ܼ���ѡ��ԭ����ҪӦӦҩ��������ܳ��������ܽ��Ҳģ��ܼ�����Ҳ��Ӧ���Ųⶨ��
(��)����ҩ��IJ�ҩ���������
���ڷ�ĩ״�ң����Խ��ⶨ��ҩ��(drug-loading rate)���Դ���Һ̬�����е��ң��ɷ����Һ���вⶨ���ټ�����ҩ���Ͱ�����(entrapment rate)��
�ⶨһ��������ĩ״���ڵ�ҩ������ҩ��������ʽ��ã�
�ҵ���ҩ����(���ڵ�ҩ�����ҵ�������)��100�� 6-7
Һ̬�����е��ң��������Ļ��˹��ȷ��������Һ�ȡһ���������ң��ֱ�ⶨ�����������ڵ�ҩ������ҩ������ʽ6-7��ã������ʿ�����ʽ���㣻
�����ʣ�[���ڵ�ҩ����(����ҩ��+�����е�ҩ��)]��100�� 6-8
���ڵ�ҩ��ռͶҩ���İٷ��ʳ�Ϊҩ��İ�����ʣ��������ҵ��������岻���������۹��ա�
�ҵİ�����ʺ���ҩ���ߵ�ȡ���ڲ��õĹ��ա���������Ϳ������������Ƶð������95�����ϵ��ң�����������뷨�Ƶõ��ң��������ʳ�Ϊ20��-80����
(��)����ҩ���ͷ�����
Ϊ����������ҩ����ͷŹ��ɡ��ͷ�ʱ�估��Ч��λ��������ҽ����ͷ����ʵIJⶨ�������ҵ��ص㡢�ɲ��á��й�ҩ��>1 995��������¼�F���Ȳⶨ���еڶ���(����)���вⶨ����ɽ������ñ�Ĥ�����ڰ���һ��(ת����)���вⶨ����������������ɲ������ط��ⶨ��
�����
[1]������� ���������������涡-��-����������������� �й�ҩѧ��־��1996��31(8);479
[2] ½��ȣ� ��Ƥ�ӷ�����-��������������о�������ҩѧ��־��1991��8(1)��10
[3] ������ȣ�ŵ��ɳ����-��������������о����й�ҽҩ��ҵ��־��1994��25(10);445
[4] �����ȣ�άA�ỷ������������Ʊ����й�ҩѧ��־��1995��30(8)��472
[5] Jimenez MC��et al.��Photodecarboxylation of 2-henylpropionic acid in so1ution and included within ��-cyclodextrin.Tetraheron�� 1995�� 51(10)��2953
[6] �ν� �����ȣ�������-��-�������������ȶ��Կ��죬�й�ҩѧ���1997��32(4)��216
[7] ��ΰ�ŵȣ���������-��-��������������Ʊ������ȶ��Եij����о����й�ҽҩ��ҵ��־��1996�� 27(11);500
[8] Erden N��Celebi N�� A Study of the inclusion complex of naproxen with ��-cyclodextrin��Int J Pharm�� l 988��18;83
[9] ����Ӣ �����壬 ���ᱽ������-��-��������������������Ƽ��������й�ҩѧ��־��1997��32(4); 218
[10] ��Ұ�����飬 ��-���ԥ��������-�������ǥ����ȥ����Ӥˤ�𥯥��ߥץ�ߥ�ι⻯ѧ��Ӧ�Ա仯�� ҩѧ��־(��)l989��109(2);107
[11] Amididouche D.et al.��lnclusion of retinoic acid in ��-cyclodextrin. Int J Pharm 1989��54(2);175
[12] �Է�Ӣ ½��˫Ȳ��̼��-PVP����������о���ҩѧѧ����1989��24(3)��219
[13] ½�� ¬������������-PEG 6000������о���ҩѧѧ����1992��27(3);227
[14] ���ű�־�� Preparation of solid dispersions of disopyramide with several coating agents and its evaluation.ҩ��ѧ(��)1992��52(1);32
[15] Murthy KS. et al.��Current perspective on the dissolution stability of solid oral dosage froms.J Pharm Sci.1993��82(2);113
[16] ½�����ȣ���Ī��ƽ���Ҷ���������ɢ����Ʊ����������ܳ��ȵ��о����й�ҩѧ��־��1995��30(1);23
[17]½��ȣ� ˫Ȳ��ʧ̼������ij������ƣ��й�ҽҩ��ҵ��־��1990��21(9);396
[18]����� ��ԪӢ�ȣ��һ���ά����Ϊ������ᶡ���������ɢ��������о��� �Ϻ�ҽ�ƴ�ѧѧ����1995��22(6)��459
[19]�Ժ� ֣����ȣ�������A�����ɢ��һЩ�����о���ҩѧѧ����l 997��32(10);777
[20] Dzeki T. et a1.��Application of the solid dispersion method to the Controlled release of medicine V. Chem Pharm Bull��1994��42(2);337
[21] Oth MP��Et al.��Sustained release solid dispersions of indomethacin with eudragit RS and RL��Int J Pharm 1989��55;157
[22] Donbrow M��Microcapsules and nanoparticles in medicine and pharmacy�� Ed��By Donbrow M��CRC Press Inc.U SA��1992��
[23] LuBin��et al.��Microncapsulation of Drugs��Ed��by whiteley TL.Harwood Academic Publishers.U��K��1992;103
[24] Fukumori Y��et a1.��Design and preparation of ethyl cellulose microcapsules of gadopentetatc dimeg1umine for neutron-capture the rapy using the wurster process�� Chem Pharm Bull��1993;41(6);1144
(½ ��)