������ע��������ժ�ԡ���������ͼ��ݡ����飻�����ص�ժ¼ҩ������Ӧ���뷢չ�������Ҳ���������ϡ����������������ݡ����շ�����ժ¼�ˣ�����뷨�������۷��������۷����ܼ�-���ܼ������֣�����������ЩͼƬ���Ʋ����ʡ�ԣ������DZ��ӵģ�
ҩ���һ�������������뷨�뵥���۷�
½��ҩ���¼������¼�����1998��171-177
һ������뷨
����빤�����ѳ�Ϊҩ���һ�����Ҫ����֮һ���������豸���߷��Ӳ�����Դ�㷺���ɽ���������ҩ���һ�����Ȼ��֪�����С��Ĥ�Ķ���ԡ�Ť���ȡ������ṹ�ȶ���������Ӱ�죬���ù��������漰һЩ����������δ�����������о�������ȷ���ۣ����ձ���ڵ��һ���ճ�����ۼ�����(����������ˮϵͳ��Ϊ����)�����ѷ��������Ŀ������ƵĹ��յõ��IJ�Ʒ��������Χ���ͷ����ݷ������źܴ�IJ��죬��˵�������ǹ��չ��̿��Ʋ��ϣ����������ڷ�Ӧ�������У�������С�仯���������Ե�Ч�����졣��ͼ��������̬ѧ��Ĥ�ṹ���о������Ѹ��㷺�����ˡ�
(һ)���� ����뷨����ҩ������ϵĻ����Һ�У��������������ʻ����ܼ�������������ʵ��ֶ�ʹ���ϵ��ܽ�Ƚ��ͣ�����Һ�в���һ������(������)�������Ʊ����ķ�������Ϊ����뷨�����ɷ�Ϊ�����۷��������۷����ܼ�-���ܼ����ı��¶ȷ���
(��)����ԭ�� �����۷�ϵ��һ�ָ߷��Ӳ���(��������CAP)�������ۼ�(�����ܽ��)ʹ֮���۳��һ������ʱ������Ϊ��ճ�ȡ��������������������ǿ���ģ�������۵��������ͷ��������۶�ʹ����ʧ���Ʊ�ʱ���������ֿ����Է������������õ����������ɹ̻����͡�
���ž۷�ʹ�����ִ��෴��ɵĸ߷��Ӳ�����Ϊ���ϲ��ϣ��������밢������(��CMC��CAP�ȶ���)������������������ᡢ���������������Ƕ��ǡ�������������ס������밢�������ȡ�
���������밢������Ϊ����˵�������۷��Ļ���ԭ��������ҺpH���������ĵȵ������ʹ֮������(pH4.0-4.5ʱ����������������)���������������Դ����磬���ڵ�ɻ����к��γ����������ӵ������ܽ�Ƚ��Ͷ����۳��һ����
���õ����ۼ������۷��õ��������֮ǰ����ʱ���������¶Ȼ��ˮϡ�͵ȣ���������������ˮ�ԣ������������ճ�ȣ�����߽���������ʹ����״�����ҿɼ���ճ����Ȼ���ٹ̻�����������ȩ��̻���CAP�ɼ���̻����������μ�CaCl2�̻��������ʿɼ��Ȼ���ȩ�̻���
�ܼ�/���ܼ����ڲ�����Һ�м���һ�ֶԲ��ϲ��ܵ��ܼ�(���ܼ�)����������룮����ҩ��������һ����ҩ������ǹ����Һ�壬��������ܼ��ͷ��ܼ������ܽ⡣Ҳ����Ӧ��ʹ����ˮ���ϣ�Ҫ���л��ܼ��ܽ⣬��ˮ��ҩ�������ϻ���ܽ⣬��ҩ������ˮ�ģ��������л��ܼ����ɻ����ڲ�����Һ�С����������л��ܼ��ķ��ܼ�ʹ���Ͻ����ܽ�ȶ�����Һ�з��룬�γ���Ĥ�����ˣ���ȥ�л��ܼ������һ���
����뷨�漰�Ļ��ư�����
1�������ܵĽ��͡���ʪ������ Һ̬ҩ�����黯��״̬��ɢ���߷��Ӳ������黯�����ã����ͽ��������Ӷ����ͽ����ܣ��߷��Ӳ��ϱ�Һ̬��������������̬������Ӧ���Ҳ��С�����������ʹ������������������������ʪ��������Ϊ�ˣ���ʱ�ɼ�����ʪ����
2����ˮ(�����ܼ�)���������� ������ˮ�����ۼ��������Ҵ����������������ǿ����ˮ���л��ܼ���Ҳ������Na2SO4��(NH4) 2SO4��A12(SO4) 3��ǿ����ˮ�����࣮���Ƕ���ʹ�߷��Ӳ�����ˮ�е��ܽ�Ƚ��͡�
3���̻� ���ݸ߷��Ӳ��ϵĻ�ѧ���ʣ�������ͨ�����Ȼ�������ѧ��Ӧ�̻�����(��������)�������۷��γ���������������������ټӽ������̻����ܼ�/���ܼ����ڳ�ȥ�л��ܼ����̻����͡�Һ̬�߷��ӿɶ���ɲ���̬�־�̬����������������ˮ���ڣ��߷����нϴ�Ļ��ء�����ʧȥ�ܼ����γ���ճ���Ե�Ĥ��������������ϣ�Ĥ���̻���������Ӧ����Ӧ�����Ĥ�Ŀ���ǿ�ȣ�����ʹĤ�γ��ѿڻ��϶����Ĥ�ĸ�����С��Ӧ������������������γɴ��ɿն����̻�������ʹĤ�״��ѣ���ͨ���̻�����Ӧ���Կ��ơ��ʵ�����ۺ����Mav�������Ĥ�Ŀ���ǿ�ȣ������ʵ������ܼ������߾ۺ���Ĥ����˳�ԡ����ܼ����γ�Ĥ�ľۺ�����Ӽ�Ӧ�нϺõ������ԣ�ͨ���������ܼ�(�������ѡ��ǻ�������)�����ڼ��Ծۺ���(����ά��������۱�ϩ����֬��)���Ǽ������ܼ�(������)�����ڷǼ��Ծۺ���(�����ۺ���)��

(��)���� ����뷨�Ƶõ���һ��������ΧΪ2��250µm��ȡ�����γɵ����������������ֲ������õĹ��ա����������뷨�Ƴ���ҩ����ҩ�������ջ������ķ�������հ���
����뷨�Ļ������տɹ���Ϊ������
1���ڸ߷��Ӳ�����Һ�У���ҩ���ܽ���ɢ�ɻ���Һ����״Һ��
2��������ǿ������¶ȡ�����pH�������ˮ�������ܼ������ۼ����Խ��߷��ӵ��ܽ�ȡ�ʹ�߷��Ӳ��ϴ���Һ���������γ��µ�����������Һ���еĸ߷��ӳ������������ϣ�����չ��Ĥ�γ��ң�С����Ҳ��Ƕ������֮��(ͼ5-2)��
3���̻����һ���
��ʵ�ϣ������������Ǿ��Է���ģ����ǻ��ཻ��ģ���ʵ�ʲ����������������ʵIJ��죬�佻��ij̶�Ҳ����ͬ����֮ҩ�P�߷��ӵ����ʵIJ��죬ҩ�������Һ̬���̬����̬ҩ��������ۻ���δ�ۻ��ģ������Ʒ�������ֲ�����̬�����Ƕ��ֶ����ģ���ͼ5-2����������ײ��Ĥ�����۹��̳������������������ֶ����ԡ���ײ���Է�������Ĥ��������֮�䡢��������Ŀ���֮�䡢��������֮��ȣ���ײ�ֿ��Է�����Ĥ���۵ij��ڡ����ڻ���ڣ���Ĥ��ճ���������ȫ�����У���ʼ���ɵ����ֿ��ڲ��Ͻ����������ٷ�ɢ���ٺϲ��������IJ�ͬ���ʹ��ʹ�����γ����Ρ���Բ�Ρ��������λ�ճ���εȣ��ҿ����Ƕ���ճ�����������ҡ�һ�Ҷ�ˡ����ҵ��ˡ�������ϲ��������Һϲ��Լ����ҵȵȡ������û����ô���ӣ���Ҳ����ֺϲ���ճ�������εȡ�
���������۷�
(һ)�Ʊ������еĽ��������븽���� �����۷��Ĺ����У����������ɢ����(�����)���䳣�õ��Ҳ���������CAP��EC��CMC���������εȣ��������ۼ�ʹ�߷����Ҳ��γ������࣬������Ӧ���������нϴ�ĸ������������һ�������ɡ�ƽ��ʱ���棬���ֽ����������Ĺ�ϵ��ͼ5-3��Ӧ�У�
![]()
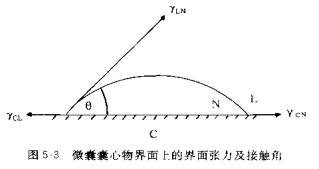
ʽ(5-1)�������ʽ(3-1)���ƣ�ֻ�ǽű겻ͬ���ű�C��ʾ�����L��ʾ��Һ��y��ʾ�����ࡣҪ��������ȫ��չ���������ϵ������ǣ��Ӵ�����=0�㣬����CL����CN+��LN��ʵ���ϣ�ֻҪ�����������������൱����������ʹ
90�㣾����0��������Ҳ���������������ʪ����չ�������������ֲڲ������¶�ʱ�����ڽ����ܴ��������ճ�Ƚ��ͣ�����������ʪ�����������ϵ����ǻ��һ�����͡�
�ڳ���ϵͳ�У�����ֻ��ҩ��������ˮ���ࡣҩ�������ˮ���ױ�ˮ������ֻ������ˮ���ж����ܻ������������г��ң�����ۻ�轺�������ﶼ�������ˮ�����ܳ��ҡ���ҩ�������ˮ�����������к�������ˮ��ʹҩ��Ȳ��ܻ�����ˮ���У��ֲ��ܻ������������У�Ҳ���ܳ��ң���˫Ȳʧ̼����������Span 20������˫Ȳʧ̼������ˮ�ԣ��Ϳ��Գ��ҡ�ҩ�����ˮ���ʵ�������������ɰ��ң�������Tween80��ʹҩ����ˮ�Թ�����˲��ܳ��ҡ�
���������ܴ������(����500mJ��m2)�������Ա����е�Һ����ʪ(���ͽ�����)��Ҳ����ͨ�����Ի�������������֬�����γ��µ���ˮ���档����������С������һ�㶼����ˮ�Եģ����ױ�ˮ��ʪ������������л���;ۺ�������Կ����ʻ�ʯ�ۼ��������踽���ڽ���������С��������ʱ������ߺ��ߵĿ���ʪ�ԡ�����̿�ܲ��ױ������/�������еľ۱�ϩ��������������ʪ(����90��)������õ��ſ飬ʹ�һ�ʧ�ܣ����춡ϩ/����ૣ��������еľ۱�ϩ�������������෴������ʪ����̿���Ӷ����Ʊ�����̿�ҡ�
��������ˮ��Ľ�������Ӧ��С��ʹ��������γ�С����״Һ�Σ���CAP������ʱ����Na 2SO4�����ۼ������Һ���������ˮ��Ľ��������ϴ����β��á��������¶��Ҽ���ˮ�Խ��ͽ��������������Ը������Ρ��������������۳��ң�������������ᣬʹ������
[174]
�и����-NH3+���������������ˮ�ԣ�������������ˮ���Ľ������������ɸ������Ρ�
������Ӧ�����ʵ��������ԣ����DZ�֤�������õ���Ҫ���������ϼ����ڽ��ͽ���������ͬʱ��Ҳ����������������ԡ���������ճ�ȳʷ��ȣ���ճ������Mav�ĺ�����ͨ��Mav������ͬ��������ճ��Ҳ����ճ�������¶ȵ����߶����͡�CAP�����۳���ʱ�轫�¶ȿ�����70-80�棬ʹ����������ʵ��������ԣ��������ҡ����Ƚ�CAP��Mav�ּ�����Mav�ϵ͵�CAP55�漴�ɳ��ҡ�
�Ҳľۺ����ڼ����ܼ���Ǽ����ܼ��ж��ɱ�������������������(��һ���¶�����������������Ũ�ȱ仯������)�ڵ�Ũ���������ܿ죬Ȼ��ﵽƽ̨�����ٻ���������ƽ̨���൱���м������Ӻ�Ľ������ھۺ����Mav�ֲ��Ͽ�����������������������ͷ�������(������)������Բ���ġ��ڲ����ܼ��У��ۺ��������������Mav����������ߣ������ܼ�����仯�����������¶�Ӱ�첻�����������ʱȵͷ�����������Ҳ������ϡ�ͽ�����
(��)�����ȶ��� ���ڷ��ù�����(�ر���������ע��Һ��)�ᷢ�����Ρ�ճ�����ۼ��Ȳ��ȶ����������Ե����۷��Ƶõ���ע��ҺΪ��������һ�ּ�������ⶨ����̬�������仯�ķ���[2]��
��Ȳŵ��ͪ(LNG)��ƶ���(E2)���ȣ��ӵ�������Һ�л������ȣ�����������ҺΪ���ۼ��Ƴ��ң���״��ͼ5-4(a)��������l0-40µm��ռ��������95�����ϣ�ƽ�������Ϊ20.7µm��
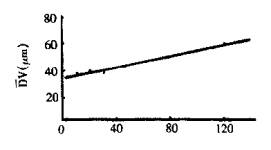
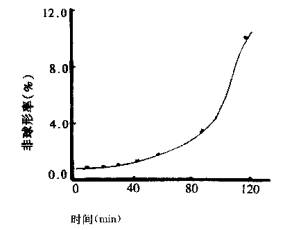
�����ȶ���������ü������顣ȡ�����һ���ע��Һl0֧���÷�ˮԡ�м��ȣ�ÿ����ʱȡһ֧���ڹ�ѧ�����¹۲�����ۣ�����(����������������)������������ҵ������(Dv)���ü��Ȳ�ͬʱ���ƽ����������������ʣ���ͼ5-4(b)(c)��
��ͼ5-4(b)�ɿ�����ƽ�������Dv��ʱ���ֱ����������ֱ��б�ʵ�Dv�仯���ʣ�0.82�������ӣ���ͼ5-4(c)�ɿ�����������Լ90����ʱ�ﵽ���ε�(�����������ߵĽ���)���ҵķ������ʿ�ʼ������������ʱ��������ԭ��������74������Ϊ���ε㣬120����ʱ���������Ѵ�10����150�����о�����֣����е��������ѡ�
������ע��Һ�ֱ���-4�桢37�漰���·��ã����37���������仯���ʣ�6.8�����£��ﵽ���ε��ʱ��Ϊ11���£����¼�-4��ﵽ���ε��ʱ��ԼΪ9-10�꣬˵������ע��Һ�������³������档
(��)ʵ��
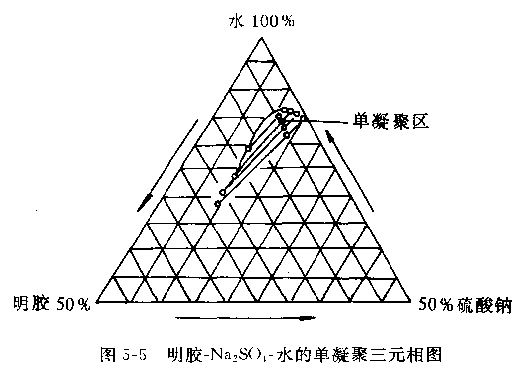
1������Ϊ�Ҳ� ����-Na2SO4-ˮ�ĵ�������Ԫ��ͼ��ͼ5-5������Ϊ�Ҳĵĵ����۷����������Ʊ���˾ƥ�֡��ǰ������Ŷ���������������ͪ���������ͪ����Ȳŵ��ͪ��˫Ȳʧ̼��������ƶ������ƶ�����Һ״ʯ��������͵�ҩ�
�����������£���ҩ����30-50g/L������Һ�����ɻ���Һ��O��W����״Һ������50�����ϡ�������pH 3.5-3.8���������ۼ�Na2SO4��Һ�������ҡ�����ϡ��Һ*�ó����ҡ�����l 5�����¼Ӽ�ȩ��Һ����NaOH��Һ����PH8-9���ù̻���(���ڲ����ü��Խ���ʱ���������ȩ����ͪȩ�ȵ�������Һ�̻���) ����ˮϴ����ȩ���ҡ�[ע��*ϡ��Һ�䷨��ϡ��Һ��Na2SO4��Һ����Ũ����������ϵͳ�е�Na2SO4Ũ�ȣ���Ϊa%��1.5%[�ã�a+1.5��%]���������Ϊ������ϵͳ�������3����ϡ��Һ�¶�Ϊ15�档����ϡ��ҺŨ�ȹ�����ͣ���ʹ������ճ�����Ż��ܽ⡣]
|
|
|
|
�仯ǰ |
�仯�� |
|
�仯ǰ�� |
|

|
|
|
|
�Ҵ�С�ı仯���� |
�������ʵı仯���� |
|
ͼ5-4 ������Ȳŵ��ͪ��ע��Һ�����ڷ�ˮԡ�еı仯 |
|
���������۷��õ��Ҽ�ͼ5-4(a)�Ĺ⾵�仯ǰ��ͼ������Ȳŵ��ͪ���ƶ����İ����ʷֱ�Ϊ68.71����74.85������SCMCΪ��������NaClΪ�����������ᱽ��Ϊ�־������Ƴɹ���ע�ĸ�����Ȳŵ��ͪ���ƶ���(��5�U2������)��ע��Һ�������lml��ƿ�У�������á�ע��Һ���Ƚ���Ϊ134.4kj��mo1��˵����ѧ�ȶ��Ըߣ������ܳ�t1/2(��Ȳŵ��ͪ)
|
|
G
|
|
|
��a����Ȳŵ��ͪ�ң���100�� |
|
|
|
(b)Ũ״ʯ����(��256) |
|
ͼ5-6 ������������ |

�о�������Ӱ���Ҳĵ����۵���Ҫ������Ũ�ȡ��¶Ⱥ͵���ʡ�Ũ��������������̫�Ͳ��ܽ������¶�������������Ũ�����ߣ��ɽ������¶��������ߣ�������е������ӶԸ߷��ӵĽ�������Ҫ���ã������������У�SO42-�ٽ�������������ǿ��C1-��֮����SCN�������ֹ��������Mav���������״���Һ�����ۣ����������������ۼ�ʱ��������Һ���������ۣ���pHֵ�أ������л��ܼ�(���༰��������)�����ۼ�ʱ��̼��������������������(��������)�������������������ۼ�ʱ���һ������ѣ�����������������������෴��ɵ����һ�������ɵĻ�����������������ͬ��ɵ���������һ�������ȡ��������ͬ�����������������ǿ�Ļ����һ�����֮ǰ���������������������ģ��ױ�������
2��CAPΪ���� ��CAPΪ���ϵĵ����۷����������Ʊ������谷���������������Ʊ������谷��Ĺ����������£�

��CAP����Na2HP04��Һ�γ�CAP-Na�������谷������Span20��ʪ�����CAPNa��Һ�С�����70��-80 ��������ۼ�Na2S04��Һ���۳���������ϳ���ϵͳ�����2-3����ϡ����̻�����pH3-4��ˮϴ����Na2S04������
CAP�����������谷��(ͼ5��7)��
3��CMC���Ҳ� CMC���Ҳĵ������Ʊ�������ҵĹ�����������[3]��
��CMC���ڷ�ˮ�У��������£��������ɻ���Һ�������µμ�A12(SO4) 3��Һ���������ҡ����ˣ�ˮϴ��80 �������ҡ�
��������������ҩ���ʹ�ϵ�ܴ���������ұȽ����С���������ԡ��ҲĩU������Ϊ1�Ul�Ƶõ��ң�������840µm��476-840µm��247-476µm�����˹�θҺ��(������Ϊ�˹���Һ�е�)��ҩ��t1/2���ֱ�Ϊ193(20)���ӡ�68(��15�����ӡ�50(��l5�����ӡ�
4�������������Ҳ� ��ˮ��Һ���Ʊ�ˮ���Բ�ͬ�ļ���ҩ��(����ҡ�������ľ�Ӹ����ѡ�������ma�Ƽ�)�ĺ��������ң���ҩ��0.51g���ڻ�����ں���������Һ��(100m1����2g�������ƣ��ɼ����ܼ�)����Ҫ�γ����ݡ���ע�����ӵ������40ml��l0g/L CaCl2�У�1-2���Ӻ�(����ʱ��̫����ˮ����ǿ��ҩ�����ˮ���������ҩ��)�����γɵ����������ˣ���ˮϴ�ӡ�60����ո���12Сʱ��ҩ��İ�����һ����80��-98%��Χ�ڣ���ҩ��24.5��-29.4������������ma�Ƽ����������������������ܼ�������ۺ��������״��ѡ���ҩ����ͬҩ���ˮ���ԡ���ҩ�����й�[4]��
�����
1��Brannon-Peppas
L.Int J. Pharm��1995��11 6��l
2��Bin Lu et a1��In Microencapsulation of Drugs ed.by whateley TL��Britain��Harwood Academic Publishers,1992��103-121
3��Ertan G��et a1��J Microencapsulation��1994��11(2)��l 27
4��Bdmme r R and wang J.J Pharm Sci��1993��82(2)��191