������ע��������ժ�ԡ���������ͼ��ݡ����飻�����ص�ժ¼ҩ������Ӧ���뷢չ�������Ҳ���������ϡ����������������ݡ����շ�����ժ¼�ˣ�����뷨�������۷��������۷����ܼ�-���ܼ������֣�����������ЩͼƬ���Ʋ����ʡ�ԣ������DZ��ӵģ�
ҩ���һ��������������۷����ܼ�-���ܼ���
½��ҩ���¼������¼�����1998��177-183
���������۷�
�����۷������������෴��ɵĸ߷��Ӳ��ϣ����ཻ���γɸ����Ҳ�����Һ������(�ɽ�ҩ���������)��������������������Ϊ���ϣ�����ˮ�������������������ߵ��������������Ĺ�ϵ����ͼ5-8��Ԫ��ͼ˵����ͼ��KΪ���������������γ��ҵĵ�Ũ�������Ͱ������������Һ��PΪ�������������������������Һ���ܻ�������γ��ң�HΪ��������������Һ�ɻ����γɾ������Һ����A�����10��������10������������80��ˮ�Ļ��Һ�������ˮϡ�ͣ���A��B���߽���������K���ܷ������ۡ�
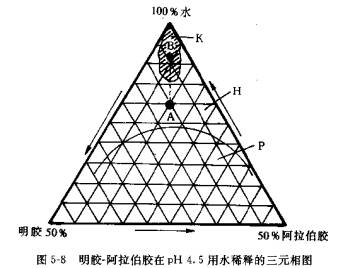
��ͼ˵��������ͬ������������������ʱ����pHֵΪ��Ҫ�����⣬Ũ��Ҳ����Ҫ������
������-��������Ϊ�Ҳĵĸ����۷������������£�
����̬��Һ̬ҩ����25-50g��L������25-50g/L����������Һ��ϣ��û���Һ��O��w����״Һ����50-55�����50g��L���ᵽpH 4.0-4.5���γ������ҡ��ó���ϵͳ���1-3����30-40���ˮϡ�ͣ����ͽ��������ó����ҡ���10�����¼����ȩ��Һ����200g��L NaOH����pH 8-9���ù̻��ҡ�ˮϴ����ȩ�����ҡ�������Ȳŵ��ͪ������ƶ����Һ��ȱ������ң��ֱ��ͼ5-9(a)[5]��(b)��
��˾ƥ�ֵ�����-�������������������ü�ȩ�̻����䶳������ҷۣ�����������2-50µm��ƽ�����ʿɴ�91.4%[6]��Ҳ�������Ʊ�����-����������������ʱ������۱�ϩ����֬�����ܼ���������Ĥ�Ŀ�϶���Ϻõؽ������©ҩ����[7]���ڰ����Ƭʱ�����ֱ�Ƭ�����������������������ι̣������ð����������Ƭ�Ƴɳ��飬��������γ���Ĥ�ٽ�����Ч���Ϻ�[8]��
����-���������������ҿ���ˮ���Ե�̼���ǰ������������Ȼ�ʹ�Ȼ����������۱�ϩ����֬���ӣ�ʹ�ұ������ʸı䣬��ǿ��θճҺ��ճ���ԣ��������ҩ���������Ч�Ժ���Ч���ø����۷��Ƶû���������-����������(�����ȩ�̻�)������1-(3-���װ�����)-3-�һ�-̼����֫(EDC�������������γ�������)����14C-�ʰ�������������Ʒ�Ӧ���ֱ�ⶨ�ұ�����Ȼ��ܶȺͰ����ܶȡ������Ȼ��IJⶨ��Ӧʽ����(Wn��ʾ��)��
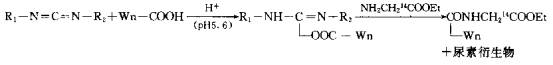
|
|
|
|
��a����Ȳŵͪ/����ƶ����� |
ͼ5-9 ����-���������������ң���100�� |


�������������ȩ����ʹ�ҵİ������١����հ�����۱�ϩ����֬��Ϻ��Ȼ�������67��(��ѱ���Ϊ�ҩU�۱�ϩ����֬������Լ6�U1)������������ʹ�հ��Ҷ�ճҺ��ճ�����Ը��ơ���
�������Ρ�CAP��SCMC�ȶ���ǻ����ͬ��������һ��������-COOH��-COO-���Ǵ�����ɵĸ߷��Ӳ��ϣ����������������������Ʊ��ҵ��Ҳġ����⣬�������ڱ�����������������-�����������������������-����������������Ҳ�����������۷��ĸ����Ҳģ����Ҳ�����������Ƕ���-�������λ�������-���������������۷������Ҳĵġ�
�Ʊ������������Ƕ���(CTS)-���������ң���10g��L������Һ�м��벻ͬ����CTS �Ƴ�1-48g��LCTS��Һ������4Сʱ��������Ĥ��ȥ�������ӣ�ȡ������Һ����NaNO2���ﵽNaN02��CTS��Ħ����Ϊ0��05�U1�����ù�ҹ(Ŀ���ǽ���CTS��Mav)��ʹ�����ϼ��ķ�Ӧ��ȫ��CTS��Mav��ԭ����1.25��l06������0. 25��l06���ֱ�ȡ�˵�Mav��CTS��Һ����һ����Mavĩ���CTS��Һ������CaCl2��1.5��������2�������ϣ�ʹ��ȫ�ܽ⣬��NaOH��Һ����pH5.5���˻��Һ��Ϊ����Һ���������°���(������Mav��CTS������ͬ)����l 5-25g��L�������Ƶ�������ˮ��Һ�У�����СţѪ�����BSAʹ���������Һ�к������Ƹ�����ȡ���10ml����Ͳʢ15g/L����������Һ10m1������22����ͷ���ÿ�����ѹ��ʹ�����������ʴ�32.0ml��h����Һ����6cm������50ml�����еİ���Һ(���轰ǡ�������Һ����)����Ӧ10-45���ӡ��������Ƶ�����500-600µm���ң���������ˮϴ�ӱ��á�������Ϊ����BSA�Ļ�BSA����CTS���ң�������15g/L��20g/L��25g/L���������Ʊ������ַ�Ӧʱ��l0-45���Ӷ�BSA���ͷ���Ӱ�죻����������Ũ�Ȼή���ͷ�BSA������(4Сʱʱ��15g/L�������ε��ͷ�37������25g��L���ͷ�20��)����Mav��CTS�Ƶõ����ͷŽϿ�(4Сʱ��77��)��δ����Mav���Ƶõ����ͷŽ���(4Сʱ��37��)���ɸ�Mav��CTS�Ƶõ������ͽϺ�(t1/2Լ8Сʱ)���ɸߵ�Mav������Ƶõ��һ�������������ʼ4Сʱ���ͷ�20����24Сʱʱ���ͷ�44�����ͷŽ��ʵ�pH�����ӿ��ͷ�(24Сʱʱ��pH3.0ʱ�ͷ�15��,pH8.0ʱ�ͷ�73������ǰ��Ĥ�����ͽ���)�������ڷ�ʱ��θҺ�л������ԣ����ڳ�Һ���ͷŽϿ�[10]��
�о����ð���-�����Ḵ�����Ʊ��������ң������临���۵�Ũ�ȷ�Χ��խ�����������ճ������Ϊ���������һ�������-��������(��pH3.9������ǿ��1mmo1/Lʱ)�ֱ������ɺ���ɣ��ҵ������ȣ���pH��Ϊ�統��pH(electrical equivalence pH��EEP)������ǿ�������ߵ��Ƶľ���ֵ�����ͣ�pH3.9ʱ����ǿ��Ϊ10mmol��L�����۵IJ������(89������1����1�U1�����)��������pH3.9ʱѸ�ٷ����ճ�Ⱥܴ��������(ճ�ȱ�����-��������������ϵͳ����߲���ʱ��3��������)�����������Ʊ����ڽӽ���߲��ʵ������£�������ǿ��l0mmol/L��pH4.2ʱ��Ϊ���ɫ��ɢϵͳ���Ƶõĸ������Ҳ������ȩ����Ҳ�����ȣ����ȶ�46��[11]��
����ѧ���ۻ����۷��Ʊ��ң�Ҫ���̬��Һ̬ҩ��ı���ɱ��Ҳ�����������ʪ���Ӷ�ʹҩ��������黯�ڸ��������в������������ɢ���ң���ʱ�����ҩ�����ʼ����ʵ�����ʪ����
�ġ��ܼ�-���ܼ���
�ھۺ�����Һ�У�����һ�ֶԸþۺ��ﲻ�ܵ�Һ��(�Ʒ��ܼ�)��������������ҩ������һ��γ���Ϊ�ܼ�-���ܼ���������ҩ�������ˮ���Ի���ˮ�ԵĹ�̬��Һ̬ҩ�������Ծۺ�����ܼ�����ܼ������ܽ⣬Ҳ����Ӧ��
������Ӧ��ʵ��
1��������(������)�� �ֱ����һ���ά��(EC)������Ϊ�����Ʊ��ҡ�����������ɢ��40g Ec����ͪ��Һ�У�����Һ״ʯ���з�ɢ��O��O����״Һ��������ˮ(���ܼ�)ʹEC���۳��ң�ϴ�ӡ������EC�ң�����ҩ������ҩ������������������ɢ������ˮ��Һ�У���Һ״ʯ���γ�w��o����״Ҵ���������(���ܼ�)��ˮ��ϴ�ӵ��ҡ����������Ҿ�Ϊ���Ρ���������ĩ����ճ����50��-60����������500-840µm��Χ�����Ҳģ��������ֵ��������ҩ�����ͣ���ҩ��t1/2������
2���ٸ�ϸ���������� ����ҩ���ŨҺ��Һ״ʯ�������黯����CAP(��ͪ���Ҵ��ܽ�)�����黯�������ȷ�(���ܼ�)�̻������ġ�������ϴ�ӡ�����������ɫ��ĩ״�Ŵ���Ƭ��ͼ5-12����ƽ������12.7µm����ҩ��29.7����������95.7����
3�� ������� ������ҩ�IJ����Eudragit RS����״������ܼ�-���ܼ����Ƶá�ÿ��2.5g�������0.833g Eudragit RS�Ӳ�ͬ��(3mL��5m1��8m1��10mI)�Ҵ��Ƶ�Ũ�Ȳ�ͬ����Һ�����Ҵ���Һ����200ml 0.25g/L������֬��������25��ˮ(���ܼ�)��Һ�н������(300 r��min)30���ӣ����ˣ���ѹ����24Сʱ����ɸ��250-1410µm������к���״�ṹ���ܽ����Ժ�ѹ�����Զ���һ�������Ҳ�ͬ���ı��Ҵ���Һ��Ũ�ȣ����Ըı����ڲ��Ķ���ԣ�Ũ�ȵ�ʱ��϶�ɸߴ��������50���������ҩ���ʷ���Higuchi���̣�����������ҩ������ʱ����ҩ���ʽ�ȡ�������ڲ��Ķ���Եij̶ȡ���Ŀ�ѹ���Դ���������Ƶõ�Ƭ������ʵ[13]��
4�� ��������� ��DL-���������ҽ���������[P(DL)LG]Ϊ������ϣ����ܼ�-���ܼ����Ʊ�5-��������� (5-Fu)��ʱ����1��0g 5-Fu�����ں�10g/L P(DL)LG������Span80��30m1�ȷ���Һ�У���800 r��min���ٽ����»����μӷ��ܼ�70mlʯ����(�ȷ²��ϻӷ�)���������ɺ��ټ�70m1ʯ���ѹ̻����ռ��������¼�ѹ�����ɸ����70������������ΧΪ105-180µm����ҩ����ҩ��57��-59��������pH7.4�����Ỻ��Һ�е������ͷ�t1/2(2.3Сʱ)Լ��ԭҩ5-Fu�ӳ�1���������ҩ��Ҫ��ͨ����ɢ���е�[14]��
�����
5������½���й�ҽҩ��ҵ��־��1994��25(11)��494
6�����ݲ��ȣ��й�ҽԺҩѧ��־��1991��11(10)��461
7���������ȣ��й�ҽԺҩѧ��־��1993��13(3);126
8�������ˣ�����.�г�ҩ��1992��14(3)��2
9. Whareley TL��Microencapsulation of Drugs Britain; Harwood Acadermic
Pblishers,1992,65-80
10��Polk A��et a1��J Pharm Sci��1994��83(2)��178
11��Burgess KK
and Singh 0N��J Pharm
Pharmacol��1993��45;586
12��Donbrow M. et
al��J
Microencapsulation,1990��7;1
13.Kawashima Y��et a1��Chem Pharm
Bull��1992��40(1)196
l4.�����ٵȣ��ִ�Ӧ��ҩѧ��1993��10(3);14